一种新型咔唑二羧酸衍生物的制备方法及其应用与流程
本发明涉及一种新型咔唑二羧酸衍生物的制备方法及其应用,其属于有机合成技术领域。
背景技术:
人工模拟光合作用的最终目标是要将太阳能转化成氢能或其它可直接利用的化学能。为实现这一目的,就需要构建水分解的光电化学电池,将对水氧化催化剂的研究由均相化学法转移到非均相电极表面,从而实现水的全分解反应。
根据已报道的文献,关于分子催化剂在电极表面的修饰方法可以分为三种:物理吸附、共价键修饰、非共价键修饰。其中,物理吸附法修饰的电极,催化剂与电极表面缺乏结合力,导致这种方法制备的电极稳定性极差,分子催化剂极易从电极表面脱落;而共价键修饰方法由于需要对分子催化剂和电极表面进行复杂繁琐的修饰反应,极大的限制了此方法的实际应用。近些年,利用非共价键修饰方法制备的电极,由于其制备工艺相对简单且具有一定的稳定性,在电化学水分解方面取得了突破性的进展。
2012年,孙立成课题组采用电泳沉积的方法,将羧基化的多壁碳纳米管(MWCNTsCOOH)沉积到ITO导电玻璃表面,选用经大共轭基团芘修饰的催化剂[RuII(bda)(Py-py)2]和MWCNTsCOOH通过π-π堆积作用相结合,制备了复合电催化水氧化电极。此催化电极在1.4V偏压下实现了从中性水溶液或盐水中高效电解水产氢产氧,法拉第效率达到95%以上,电催化产氧速率(TOF)达到0.8s-1。而且此复合电极展现出很好的稳定性,经过10小时的电催化水氧化,TON达到了11000。但是此复合电极的催化电流密度只有220μA/cm2,且TOF值也很低。
2014年,孙立成课题组在催化剂[RuII(pda)(pic)3]的骨架配体上引入一个12个碳的烷氧基长碳链合成了具有强疏水性的催化剂,并利用其疏水性能与羧基化的多壁碳纳米管(MWCNTsCOOH)相结合,采用过滤的方法共同负载到具有良好导电性的碳材料基底上制备了具有电催化水氧化性能的复合电极。实验证明,此复合电极表现出了很好的电催化水氧化性能,在1.3V的偏压下,初始催化电流密度可达4.5mA/cm2,TOF值高达12.4s-1,并且可以持续工作10小时以上,法拉第效率达到96%,TON可达170000以上。但是此复合电极的催化电流密度衰减速率很快,稳定性不够好,经1小时反应后,其电流密度降为2mA/cm2,5小时后,其电流密度降至1mA/cm2以下。
咔唑衍生物是一种具有刚性稠环结构的化合物,具有很多独特的性能,在医药,染料,光电领域有很多潜在应用。咔唑衍生物的优势之一就是容易进行结构修饰,在咔唑环上引入官能团,开拓咔唑衍生物的用途。
技术实现要素:
本发明的目的在于,在咔唑环上引入两个十二烷基碳链,合成新的咔唑衍生物,开拓咔唑衍生物在染料、医药、有机光电等领域中的应用;将该咔唑二羧酸衍生物应用于有机光电材料中,以提高其作为催化剂时的稳定性。
为实现上述目的,本发明采用的技术方案是:新型咔唑衍生物,所述化合物具有以下结构:
本发明的另一个目的,提供了一种新型咔唑衍生物的制备方法,包括以下步骤:
(1)化合物Ⅱ与十二烷酰氯发生Friedel—Crafts反应,
得到化合物Ⅲ:
(2)化合物Ⅲ与四氢锂铝和三氯化铝在四氢呋喃中搅拌3h,得到化合物Ⅳ:
(3)化合物Ⅳ与溴素常温搅拌反应,得到化合物Ⅴ:
(4)化合物Ⅴ与氰化亚铜加热回流得到化合物Ⅵ:
(5)化合物Ⅵ与氢氧化钠加热回流得到化合物Ⅰ:
进一步地,所述步骤(1)中,室温搅拌下分批加入无水三氯化铝,继续搅拌反应90分钟。将反应置于冰水浴中冷却至0℃,缓慢滴加十二烷酰氯,滴加完毕后将反应温度升至室温,继续搅拌反应14小时。将反应液倒入冰水中,过滤收集沉淀,分别用去离子水和甲醇进行淋洗,收集粗产品并用甲醇/丙酮重结晶,真空干燥得白色纯品。化合物Ⅱ与十二烷酰氯摩尔比1:2。
进一步地,所述步骤(2)中,0℃下分批加入化合物Ⅲ,室温搅拌反应3小时。然后向反应液中缓慢滴加乙酸乙酯和5%的盐酸,过滤除去不溶物,旋干滤液。
进一步地,所述步骤(3)中,化合物Ⅳ和冰醋酸加入到100mL圆底烧瓶中,室温搅拌避光条件下,向反应液中滴加溴素,继续室温反应1小时。反应结束后,加入饱和硫酸硫代钠饱和水溶液,用乙酸乙酯进行萃取,收集有机相,加入无水硫酸镁进行干燥,过滤,旋干滤液得黄色固体产物。
进一步地,所述步骤(4)中,化合物Ⅴ、氰化亚铜和N-甲基吡咯烷酮加入到100mL两口烧瓶中,加热回流反应过夜。停止反应冷却至室温,向反应液中加入饱和三氯化铁溶液和乙酸乙酯,继续搅拌半个小时,过滤除去不溶物,滤液用乙酸乙酯萃取,合并有机相,用氯化钠溶液水洗,收集有机相并加入无水硫酸镁进行干燥,过滤,旋干滤液。
进一步地,所述步骤(5)中,将化合物Ⅵ和NaOH溶液加热回流反应过夜,直至反应物全部溶解。停止反应,冷却至室温,用浓盐酸将反应液的pH值调至1,发现有大量沉淀生成,减压抽滤,水洗,收集滤饼,真空干燥得到新型咔唑衍生物。
所述咔唑二羧酸衍生物应用于有机光电材料中。
本发明的有益效果为:该咔唑二羧酸衍生物的主要特点为引入了两个长碳链,该衍生物与Ru形成配合物时,骨架配体咔唑上引入的两个长碳链对中心金属Ru的配位不产生影响;同时,两个自由摆动的长碳链使得配合物Ru具有极强的疏水性,与碳纳米管之间可以很好地作用结合。以该咔唑二羧酸衍生物为配体制备的配合物Ru吸附于碳纳米管上,成功制备了复合电极Ru6/MWCNTsCOOH/GC。电化学研究结果表明,此电极具有很好的电催化水氧化活性,其过电位仅为380mV。中性磷酸缓冲溶液中的长时间电解实验证实,在1.4V的工作电压下,复合电极可以持续工作4小时电流无衰减,进一步的稳定性实验表明复合电极可以稳定工作10小时以上,催化电流密度仍保持在起始电流的80%,为1mA/cm2,对应的催化产氧速率(TOF)为10s-1,TON高达360000,法拉第效率达到96%。
附图说明
图1是新型咔唑衍生物的1H NMR谱图。
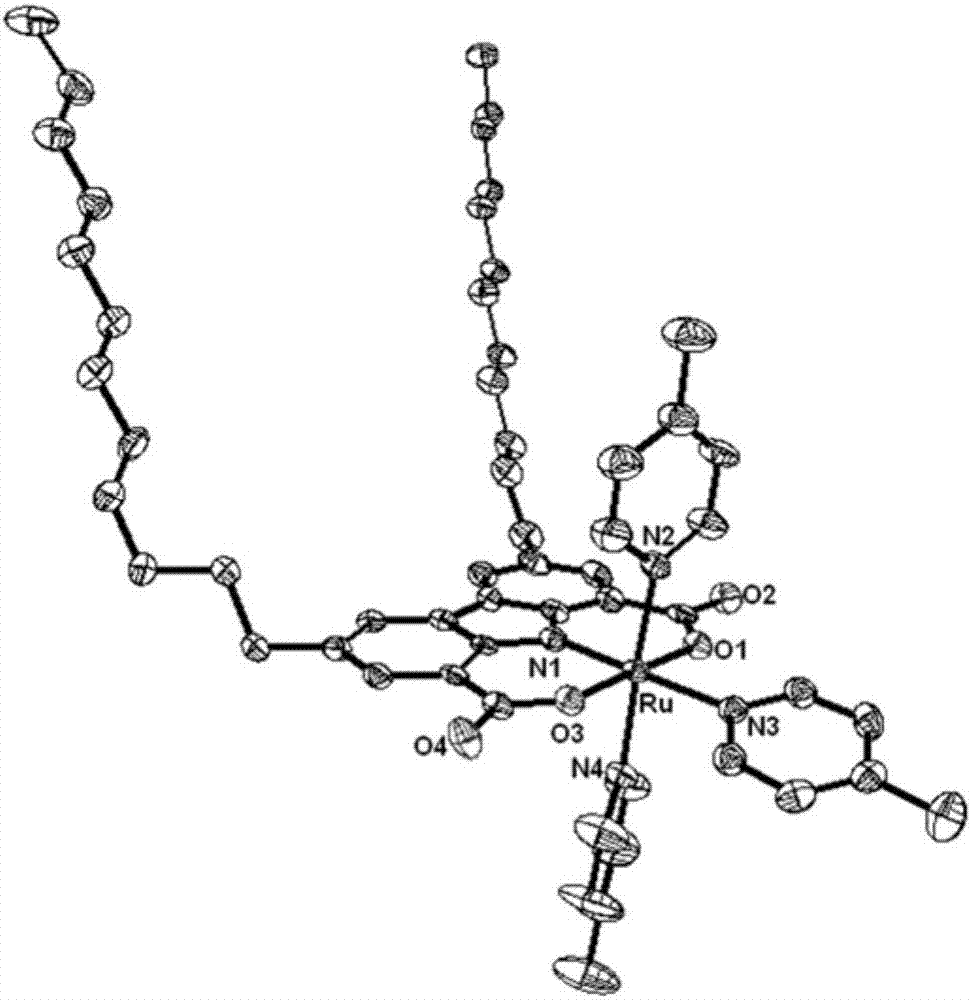
图2是配合物Ru的晶体结构。
图3是复合电极Ru/MWCNTsCOOH/GC的结构。
图4是复合电极Ru/MWCNTsCOOH/GC的扫描电镜图。
图5是电极MWCNTsCOOH/GC与复合电极Ru/MWCNTsCOOH/GC的EDX图。
图6是复合电极Ru/MWCNTsCOOH/GC的元素分布图。
图7是复合电极Ru/MWCNTsCOOH/GC的循环伏安扫描测试图。
图8是复合电极Ru/MWCNTsCOOH/GC恒电位电解。
图9是复合电极Ru/MWCNTsCOOH/GC不同电位条件下的电流密度测试。
图10是复合电极Ru/MWCNTsCOOH/GC不同过电位条件下的TOF值。
图11是复合电极Ru/MWCNTsCOOH/GC长时间电解测试。
图12是复合电极Ru/MWCNTsCOOH/GC的法拉第效率测试。
图13是复合电极Ru/MWCNTsCOOH/GC电催化水氧化的过程图。
具体实施方式
以下结合实施例对本发明进一步说明:
实施例1咔唑衍生物I的制备
向250mL两口瓶中加入咔唑(5.02g,30mmol)和100mL CH2Cl2,室温搅拌下分批加入无水三氯化铝(9.33g,70mmol),继续搅拌反应90分钟,将反应置于冰水浴中冷却至0℃,缓慢滴加十二烷酰氯(15.30g,70mmol),滴加完毕后将反应温度升至室温,继续搅拌反应14小时。将反应液倒入200mL冰水中,过滤收集沉淀,分别用去离子水和甲醇进行淋洗,收集粗产品并用甲醇/丙酮重结晶,真空干燥得白色纯品3,6-二(十二酰基)咔唑(11.20g,产率70%)。1H NMR(400MHz,CDCl3,):δ(ppm)8.98(br,1H),8.79(s,2H),8.14(d,J=8.8Hz,2H),7.49(d,J=8.8Hz,2H),3.11(t,J=7.2Hz,4H),1.90-1.80(m,4H),1.45-1.20(m,32H),0.88(t,J=6.8Hz,6H).13C NMR(400MHz,CDCl3):δ(ppm)200.12,142.85,130.14,127.05,123.36,121.65,110.81,38.66,31.90,29.63,29.55,29.33,24.85,22.67,14.08.TOF-MS:m/z=[M+H]+calcd:532.4155;found:532.4175。
将四氢锂铝(3.04g,80mmol)、三氯化铝(5.33g,40mmol)和120mL干燥的THF加入到250mL圆底烧瓶中,0℃下分批加入3,6-二(十二酰基)咔唑(10.60g,20mmol),室温搅拌反应3小时。然后向反应液中缓慢滴加100mL乙酸乙酯和5mL 5%的盐酸,过滤除去不溶物,旋干滤液得到粗产品,然后以二氯甲烷/正己烷=2:1(V:V)为洗脱剂进行硅胶柱分离,得到白色固体产物(7.16g,71%)。1H NMR(400MHz,CDCl3):δ(ppm)7.85(s,2H),7.82(br,1H),7.29(d,J=8.4Hz,2H),7.21(d,J=8.4Hz,2H),2.76(t,J=7.6Hz,4H),1.75-1.60(m,4H),1.45-1.20(m,36H),0.88(t,J=6.8Hz,6H).13C NMR(400MHz,CDCl3):δ(ppm)138.19,133.81,126.38,123.37,119.51,110.16,36.05,32.37,31.92,29.69,29.64,29.61,29.39,29.36,22.69,14.13.TOF-MS:m/z=[M+H]+calcd:504.4569;found:504.4593。
将3,6-二(十二烷基)咔唑(2.50g,5mmol)和60mL冰醋酸加入到100mL圆底烧瓶中,室温搅拌避光条件下,向反应液中滴加溴素(0.60mL,12mmol),继续室温反应1小时。反应结束后,加入饱和硫酸硫代钠饱和水溶液50mL,用乙酸乙酯(3×100mL)进行萃取,收集有机相,加入无水硫酸镁进行干燥,过滤,旋干滤液得黄色固体产物1,8-二溴-3,6-二(十二烷基)咔唑(3.20g,98%)。1H NMR(400MHz,CDCl3):δ(ppm)8.08(br,1H),7.89(s,2H),7.60(d,J=8.1Hz,2H),2.72(t,J=7.4Hz,4H),1.78-1.62(m,4H),1.41-1.10(m,36H),0.86(t,J=6.5Hz,6H).TOF-MS:m/z=[M+H]+calcd:660.2780;found:660.2798.
1,8-二溴-3,6-二(十二烷基)咔唑(2.64g,4mmol)、氰化亚铜(0.92g,10mmol)和40mL N-甲基吡咯烷酮加入到100mL两口烧瓶中,加热回流反应过夜。停止反应冷却至室温,向反应液中加入饱和三氯化铁溶液(80mL)和乙酸乙酯(50mL),继续搅拌半个小时,过滤除去不溶物,滤液用乙酸乙酯(3×100mL)萃取,合并有机相,用氯化钠溶液(2×50mL)水洗,收集有机相并加入无水硫酸镁进行干燥,过滤,旋干滤液得粗产品,再用乙酸乙酯进行重结晶得固体产物1,8-二氰基-3,6-二(十二烷基)咔唑(1.77g,80%)。1H NMR(400MHz,CDCl3):δ(ppm)9.87(s,1H),8.21(d,J=1.5Hz,2H),7.85(d,J=1.6Hz,2H),2.74(t,J=6.8Hz,4H),1.75-1.60(m,4H),1.45-1.10(m,36H),0.83(t,J=5.5Hz,6H).TOF-MS:m/z=[M+H]+calcd:554.4474;found:554.4492。
将1,8-二氰基-3,6-二(十二烷基)咔唑(1.66g,3mmol)和NaOH(1.20g,30mmol)加入盛有20mL去离子水的100mL两口瓶中,加热回流反应过夜,直至反应物全部溶解。停止反应,冷却至室温,用浓盐酸将反应液的pH值调至1,发现有大量沉淀生成,减压抽滤,水洗,收集滤饼,真空干燥得到产品1,8-二羧基-3,6-二(十二烷基)咔唑(1.70g,96%)。1H NMR(500MHz,d6-DMSO):δ(ppm)10.94(s,0.5H),8.30(s,1H),7.86(s,1H),2.76(t,J=7.2Hz,2H),1.78–1.60(m,2H),1.42–1.08(m,18H),0.81(t,J=5.4Hz,3H).TOF-MS:m/z=[M–H]-calcd:590.4209;found:590.4237。
实施例2以咔唑二羧酸衍生物为骨架的金属配合物Ru的制备
化合物Ⅰ与Ru(DMSO)4Cl2在Et3N和无水DMF中,加热至110℃反应12h。然后再加入4-甲基吡啶,继续90℃反应10小时。停止反应,冷却至室温,将溶剂旋干,得到深绿色粉末产品。化合物Ⅰ与Ru(DMSO)4Cl2的摩尔比为1:1,4-甲基吡啶是过量的。以上步骤均在无水无氧条件下进行。
最后,以CH2Cl2/CH3OH=100:1(V:V)为洗脱剂进行硅胶柱分离,得到绿色固体产物(产率45%)。TOF-MS:m/z=[M+H]+calcd:970.4910;found,970.4941.m/z=[M+Na]+calcd:992.4730;found:992.4754.Elem.anal:calcd.for C56H75N4O4Ru(%):C 69.32,H 7.74,N 5.78;found,C69.23,H 7.72,N 5.88。
实施例3金属配合物Ru的晶体培养与解析
配合物的晶体培养方法:向5mL溶有配合物(15mg)的甲醇溶液中缓慢加入0.1mL的去离子水,密封保存,室温静置五天,通过甲醇和水的相互扩散使得整个体系过饱和,析出适合单晶测试的绿色晶体。
对配合物的结构进行了X-射线单晶衍射测试,获得了其X-射线单晶衍射数据。具体测试仪器与方法见第二章2.2.3节。配合物的晶体学参数和测试参数见表1。
表1配合物Ru的晶体测试数据和相关参数
通过X-射线单晶衍射测试,获得配合物Ru的精确空间结构(见图2)。配合物Ru主要的键长和键角列于表2中,晶体的CCDC号为CCDC–1499670。
表2配合物Ru的主要键长和键角数据
通过对配合物Ru的晶体数据进行解析,与骨架配体咔唑(不带长碳链)相同的已知化合物对比,配合物Ru的主要键长和键角与其几乎一样。这表明,骨架配体咔唑上引入的两个长碳链对中心金属Ru的配位并没有产生影响。同时,两个自由摆动的长碳链使得配合物Ru具有极强的疏水性,与碳纳米管之间可以很好地作用结合。
实施例4复合电极Ru/MWCNTsCOOH/GC的制备
(1)电极MWCNTsCOOH/GC的制备
玻璃碳电极的预处理:用粒径为3μm氧化铝粉打磨5分钟,用去离子水清洗,然后超声清洗10分钟,用拭镜纸擦干备用。
MWCNTsCOOH分散液的制备:将5mg MWCNTsCOOH置于5mLTHF中,超声处理20min,得到MWCNTsCOOH的THF分散液。
用移液枪取60μL MWCNTsCOOH分散液滴涂到玻璃碳电极表面,控制面积为1cm2,待THF挥发完全,再重复此操作两次。得到MWCNTsCOOH/GC电极,放到烘箱中70℃烘干备用。
(2)复合电极Ru/MWCNTsCOOH/GC的制备
将制备好的MWCNTsCOOH/GC电极浸泡在1.0mM的配合物Ru的甲醇溶液中过夜,取出之后用0.1mL甲醇冲洗掉电极表面残存的催化剂溶液,N2吹干得到Ru/MWCNTsCOOH/GC复合电极(见图3)。
羧基化的碳纳米管相对普通的碳纳米管而言,与水分子之间具有一定的氢键作用,可以提高电极的电化学催化水氧化的活性。因此,本申请采用阿拉丁试剂公司生产的羧基化多壁碳纳米管作为负载分子催化剂的载体材料,同时选用具有良好导电性能的玻璃碳作为基底材料,并采用简单的滴涂法将羧基化多壁碳纳米管负载到玻璃碳表面,然后利用浸泡的方法将催化剂担载到碳纳米管上,从而制备复合电极,这种制备方法简单易操作,且具有很好的可重复性。此外,这种方法制备的复合电极具有可重复使用的优点,在测试完成后,对电极进行打磨抛光,再用甲醇溶液超声处理后可用于再次制备复合电极,这对于以后的商业化应用具有重要的意义。
从复合电极的扫描电镜图(图4)可以看出,玻璃碳电极表面的碳纳米管相互交织重叠,呈现出无序多孔的结构,因此具有很大的比表面积,为分子催化剂的负载提供了保障。此外,通过对比电极MWCNTsCOOH/GC和复合电极Ru/MWCNTsCOOH/GC的能量色散X射线光谱(EDX)的测试结果(图5),可以清晰观察到Ru/MWCNTsCOOH/GC复合电极上有金属Ru,证明分子催化剂确实负载到了电极上。在扫描电镜测试的同时,对电极表面的主要元素分布进行了分析(图6),在随机选取的一块区域内进行测试分析,结果显示,电极表面的C元素和Ru元素分布都十分均匀,这表明分子催化剂在复合电极表面是均匀分布的。
实施例5复合电极Ru/MWCNTsCOOH/GC的电化学测试方法和条件
选用上海华辰公司的CHI 630E型电化学工作站对Ru/MWCNTsCOOH/GC复合电极进行循环伏安和恒电位电解测试,在离子强度(IS)为0.1M的pH 7.0磷酸缓冲溶液中,以1cm2的铂网做对电极,以Ag/AgCl(3.5M饱和KCl溶液)为参比电极,所有电位均通过Ru(bpy)32+(RuII/RuIII=1.26V vs.NHE)进行参比校正。所有恒电位电解测试均未进行iR降电压补偿。
实施例6复合电极Ru/MWCNTsCOOH/GC的电催化水氧化的应用
通过对复合电极Ru6/MWCNTsCOOH/GC进行循环伏安扫描(扫描速率为100mV/s)测试(图7),可以观察到复合电极Ru/MWCNTsCOOH/GC具有很好的电化学催化水氧化活性,在E1/2为0.59V处可以清晰地观察到一对氧化还原峰,此氧化还原过程对应的是配合物Ru的RuIII/RuIV过程。通过对RuIII/RuIV氧化还原峰峰电流积分得到电量为(2.9±0.3)×10-5C,进而通过法拉第定律公式Γ=Q/(nsF)(n为电子转移数,等于1;s为电极的工作面积,等于1cm2;F为法拉第常数,等于9.65×104C/mol),可以计算出此复合电极上催化剂的吸附量为(3±0.3)×10-10mol/cm2。循环伏安测试还显示此复合电极电化学催化水氧化的启动电位为1.2V,过电位仅为380mV。而没有修饰催化剂的MWCNTsCOOH/GC电极和GC电极在循环伏安扫描范围内则没有观察到任何氧化还原峰,这说明此二电极几乎没有电催化水氧化的活性。
为进一步考察复合电极Ru6/MWCNTsCOOH/GC的电催化水氧化活性,以pH 7.0的磷酸冲溶液为电解液,控制电极的工作面积为1cm2,在1.4V的外加偏压下进行了恒电位电解实验,结果如图8所示。在电解的过程中,可以清晰地观察到大量的气泡不断地从Pt网阴极与Ru/MWCNTsCOOH/GC阳极上放出,经气相色谱检测确定分别为氢气和氧气。经过2小时的电解实验,电流密度维持在1.25mA/cm2左右无衰减,表明该复合电极对于电化学催化水氧化制氢反应具有很好的稳定性。
(1)复合电极Ru/MWCNTsCOOH/GC的Tafel和TOF测试
对复合电极Ru6/MWCNTsCOOH/GC在不同偏压下进行控制时间的电催化水氧化测试,得到不同过电位条件下的催化电流密度(图9)。然后将催化电流密度取对数作为纵坐标与过电位作为横坐标作图,可以得到复合电极的Tafel曲线(图10)。
从图10中可以看出,在380mV到780mV的过电位范围内,复合电极的Tafel呈一条直线。通过公式TOF=Q/(4tFΓ)(Q为电解过程中通过的电量,单位为C;F为法拉第常数,等于9.65×104C/mol;t为电解反应的时间,单位为s;Γ为催化剂的吸附量,单位为mol)可以计算出复合电极在不同过电位下的TOF值。随着过电位从380mV增大到780mV,电极的TOF值也相应的从2.2s-1增大至31.5s-1,这说明此电极具有很高的电催化水氧化活性。
(2)复合电极Ru/MWCNTsCOOH/GC的电催化水氧化稳定性测试
为进一步评估复合电极Ru6/MWCNTsCOOH/GC的电催化水氧化反应的稳定性,本发明对复合电极进行了长时间电解测试。从图11的测试结果可以看出,电解10小时后电极的催化电流密度仍有1mA/cm2,这一结果证明复合电极Ru6/MWCNTsCOOH/GC在展现出很高电催化水氧化活性的同时还具有很好的稳定性。此类催化剂在催化水氧化过程中生成的中间体为电中性,也就是说,配合物Ru在整个催化水氧化反应过程中始终为电中性,这就保证了配合物Ru在电解过程中不易从电极表面脱落,这一结论与电解测试的结果一致。
实施例7复合电极Ru/MWCNTsCOOH/GC的法拉第效率测试
采用天津艾达公司的可封闭式电解池对复合电极Ru6/MWCNTsCOOH/GC的法拉第效率进行测试。首先,将电解池中加入一定量的pH 7.0缓冲溶液作为电解液,再将复合电极、铂网电极与Ag/AgCl电极固定,用聚四氟胶带将电解池密封,然后向电解池中鼓入氩气进行除氧,气相检测除氧完成后,每隔一定的时间,采用手动进样的方法通过气相色谱对电解池内生成的氢气与氧气进行检测和定量,最后通过计算得到法拉第效率值。测试结果见图12。
法拉第效率是指电催化水氧化反应过程中用于产氧的电荷量与电路中通过的总电荷量之比,对于水氧化反应而言,每生成一个氧气分子需要消耗四个电荷,因此法拉第效率可以通过公式η=4nO2/Q(nO2为电解过程中产生的氧气量,单位为mol;Q为电解过程中通过的总电荷量,单位为mol)。在密闭电解池中,1.4V电压下进行长时间电解测试,同时通过气相色谱对电解过程中生成的氧气进行定量,电解10小时后,检测到生成的氧气量为107.8μmol,换算成电量为41.6C,与电解过程中通过的总电量43.1C吻合,经计算得知复合电极的电催化水氧化反应的法拉第效率高达96%,TON达到360000。与此同时,通过气相检测到生成的氢气量为217.4μmol,与氧气的比例为2:1。
最后应说明的是:以上各实施例仅用以说明本发明的技术方案,而非对其限制;尽管参照前述各实施例对本发明进行了详细的说明,本领域的普通技术人员应当理解:其依然可以对前述各实施例所记载的技术方案进行修改,或者对其中部分或者全部技术特征进行等同替换;而这些修改或者替换,并不使相应技术方案的本质脱离本发明各实施例技术方案的范围。
- 还没有人留言评论。精彩留言会获得点赞!