含磷膨胀型成炭剂及其合成方法与流程
本发明属于环保型无卤阻燃产品的技术领域,具体地说,本发明涉及一种含磷和三嗪结构的膨胀型成炭剂及其合成方法。
背景技术
高分子材料及其产品广泛应用于人们的日常生产生活,但是大多数高分子材料都属易燃材料,且燃烧有滴落产生,加快了火灾的扩展,大大限制了部分场合的使用,因此,高分子材料阻燃改性成为重要的内容。
传统的高效阻燃剂中,一般含有卤素,尤其是溴,当此类阻燃剂与三氧化二锑复配后对高分子材料具有高效广谱的阻燃效果。但是此类阻燃剂在燃烧或加工时会释放出大量卤代氢、二噁英等有毒有害物质,造成二次污染,从而严重地破坏了周围环境。
膨胀型阻燃剂(ifr)在燃烧时能形成致密的炭层,减少有毒有害气体和烟雾的释放,并能有效地防止燃烧过程中的熔滴,因此,特别适用于烯烃类聚合物的阻燃。ifr在阻燃剂领域中越来越受到重视,是一种非常有应用前景的绿色环保型阻燃剂。膨胀型阻燃剂(ifr)通常由炭源、酸源和气源三种组份构成。传统的膨胀型阻燃剂中,成炭剂主要使用季戊四醇及其衍生物、淀粉等多羟基化合物,此类成炭剂容易迁移,水溶性大,热稳定性不高,加工过程中容易与多聚磷酸盐反应,因此,新型成炭剂的开发是当前膨胀型阻燃剂研究的一个重要方向。
富含叔氮结构的三嗪类衍生物具有优良的成炭性能,引起了研究人员的密切关注和广泛开发。三嗪类衍生物的合成一般是采用三聚氯氰为起始原料。利用三聚氯氰分子结构中含有三个氯原子反应活性的差异,在不同的反应条件下可以分别发生亲核取代反应,因此,采用不同的工艺可以得到不同用途和性质的三嗪类衍生物;并且三聚氯氰的合成工艺简单成熟,廉价易得,具有成本优势。
日本专利jp.pat.no.0,583,065,al(1994)中公开了采用三聚氯氰为起始物制备三嗪类成炭剂,产率约为91%,总反应耗时长,时间在29小时以上,操作复杂。
专利zl200510010243.4《大分子三嗪系成炭-发泡剂及其合成方法》和专利《三嗪系齐聚物及其合成方法》(授权号cn100500657c),描述了一种以三聚氯氰、二胺为原料制备的三嗪齐聚物,具有良好的热稳定性、和阻燃效果,但高温下残炭量不大。
专利《含芳香族链结构的三嗪成炭剂及其制备方法》(公开号cn101586033a),公开了一种含芳香族链结构的三嗪成炭剂,采用“一锅法”工艺制备,中间体不需要处理,产率高,有很好的成炭性和热稳定性,但是需要使用大量的溶剂,并且芳香族的h2n-ar-nh2,ho-ar-oh,hs-ar-sh溶解性能和反应活性较差,需要使用强极性溶剂,如dmf、dmso等,反应温度也较高。
此外,还有一些三嗪类成炭剂的合成报道。如:胡小平等(journalofappliedpolymerscience94,1556-1561,2004)等,但是合成的三嗪类齐聚物热稳定性差,在100℃失重已经达到9%,且成炭性能也不佳。
这些专利和合成方法在不同程度地存在如下问题:产物的热稳定性、成炭性性能还有待提高,并且含有大量的极性基团,吸水性较强,易使材料吸潮。
技术实现要素:
为解决上述问题,本发明的目的是针对现有技术上存在的产物的成炭量低和热稳定性较差的缺点,本发明的第一目的是提供一种含磷膨胀型成炭剂,本发明的第二个目的是提供一种含磷膨胀型成炭剂的合成方法。
为了实现本发明的第一目的,本发明的技术方案是这样实现的,其是一种含磷膨胀型成炭剂,其特征在于,结构式如下:
式中:r1为含磷结构,其结构式为下式(ⅰ)或(ⅱ)或(ⅲ)所示:
y为n-x或o,n-x中的x为h或含1-18个碳的直链或带支链烷基中的一种;
r2为含1-18个碳的直链或支链烷基,含有氧、氮元素直链或支链烷基;
r3为苯基、带有取代基的苯基、含1-18个碳的直链或支链烷基、环烷基或带侧基的环烷基;
r4为h或含1-18个碳的直链或支链烷基。
为了实现本发明的第二目的,本发明的技术方案是这样实现的,其是一种含磷膨胀型成炭剂的合成方法,其特征在于,包括如下几个步骤:
步骤一
将溶剂和三聚氯氰加入到容器中,溶剂和三聚氯氰的比例是200~700ml:1mol;滴加r3-yh与缚酸剂的水溶液,控制ph在5-7,反应温度控制在-10-30℃,反应1-3小时,得到三聚氯氰一元取代物,其中三聚氯氰与r3-yh的摩尔比为1:1;
步骤二
在另一容器中加入hy-r2-yh与溶剂、缚酸剂混合物,温度控制在-10~10℃,缓慢滴加含磷化合物,反应3-6小时,分离得到含磷二胺中间体;其中含磷化合物、hy-r3-yh以及缚酸剂的摩尔比为1:2-5:2;1mol的含磷化合物用50-500ml的溶剂;
步骤三
在40-50℃的条件下,向装有三聚氯氰一元取代物的容器中缓慢滴加步骤二中得到的含磷二胺中间体和缚酸剂,反应2-8小时;之后,提高反应温度到80-150℃,继续反应5-10小时,洗涤,干燥,得到白色或浅黄色固体即具有权利要求1所述的结构式的含磷膨胀型成炭剂,其中三聚氯氰一元取代物、含磷中间体以及缚酸剂的摩尔比为1:1:1;
本发明的合成路线如下:
在本技术方案中,所述溶剂为水、丙酮、乙腈、四氢呋喃、丁酮、乙酸乙酯、二氧六环、二氯甲烷、三氯甲烷、甲苯、二甲苯、三甲苯、四甲苯、二甲亚砜、n,n-二甲基甲酰胺中的一种或两者的混合液。
在本技术方案中,所述缚酸剂为三乙胺、三乙烯二胺、吡啶、n,n-二异丙基乙烷、氢氧化钠、氢氧化钾、氢氧化钙、氢氧化钡、碳酸钠、碳酸氢钠、碳酸钾或碳酸氢钾中的一种。
本发明与现有技术相比的优点为:产品为白色或浅黄色粉末,产率在90%以上,具有高的含碳和含氮量,并且含有磷;该聚合物不仅是良好的成炭剂,而且由于燃烧过程中产生nh3、n2等不燃性气体,可以提供阻燃体系所需的气源,起到膨胀炭层和稀释可燃性气体的作用,同时,含有磷能促使成炭剂成炭,提高残炭量,改善炭层结构,提高阻燃性能;产品在加热时分解发泡膨胀,最终形成膨胀致密的炭层,具有成炭性和热稳定性好,炭层结构稳定。
附图说明
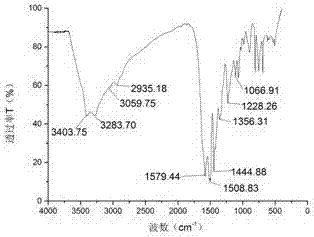
图1为本发明实施例1合成的含磷膨胀型成炭剂(cnca-p)的ftir谱图;
图2为本发明实施例1合成的含磷膨胀型成炭剂(cnca-p)的tga图。
具体实施方式
下面结合实施例对本发明作进一步说明。在此需要说明的是,对于这些实施方式的说明用于帮助理解本发明,但并不构成对本发明的限定。此外,下面所描述的本发明各个实施方式中所涉及到的技术特征只要彼此之间未构成冲突就可以互相结合。
实施例一
步骤一
在装有回流冷凝器、温度计、搅拌器、恒压滴液漏斗的1000ml四口瓶中,加入92.25g(即0.5mol)的三聚氯氰,然后加入200ml丙酮,将四口瓶置于冰浴中,充分搅拌,使三聚氯氰分散均匀,往四口瓶中滴加46.56g(即0.5mol)的苯胺,又将20g(即0.5mol)的氢氧化钠溶于50ml蒸馏水并用恒压漏斗滴加到四口瓶中,控制ph在5-7,反应温度控制在-10℃,反应时间为3小时,得到三聚氯氰的一元取代物;
步骤二
在另外一个装有搅拌器、恒压滴液漏斗的1000ml三口烧瓶中,加入120.20g(即2mol)的乙二胺、101.20g(即1mol)的三乙胺和200ml的三氯甲烷,冷却到0℃,用恒压漏斗滴加105.49g(即0.5mol)的二氯化磷酸苯酯到三口瓶中,搅拌反应4小时,旋蒸除去未反应的化合物和三氯甲烷,得到含磷二胺中间体;
步骤三
将上述含磷二胺中间体溶解到n,n-二甲基甲酰胺中,转入恒压漏斗,缓慢滴加到步骤一装有三聚氯氰的一元取代物的四口瓶中,逐步升温到50℃,反应4小时;之后继续升温到120℃,反应6小时,反应结束;洗涤、过滤、烘干,得到浅黄色沉淀(cnca-p),共188.6g,产率为95.5%。
本实施例的合成路线如下:
如图1所示,ftir分析结果为:波数为1356.31cm-1为-nh-和三嗪环上的碳相连接的-c-n-的伸缩振动,1444.88cm-1、1508.83cm-1、1579.44cm-1是三嗪环的骨架振动峰、1228.26cm-1为p=o键的伸缩振动峰,波数为3283.70cm-1是n-h键之间的伸缩振动峰,说明有新的-nh2接入;由此说明,所合成的产物与设计的分子结构基本吻合。
如图2所示,tga分析结果表明:cnca-p在空气中的初始分解温度为233℃,800℃时的残余率为8.85%,说明cnca-p的具有较好的成炭性。
当多聚磷酸铵(app)与cnca-p按质量比2:1复配时,阻燃效果最佳,当总添加量为30%时,pp/app/cnca-p复合材料的氧指数值为36.7%,通过
ul-94v-0级;cnca-p的结构式如下:
实施例二
步骤一
在装有回流冷凝器、温度计、搅拌器、恒压滴液漏斗的1000ml四口瓶中,加入92.25g(即0.5mol)的三聚氯氰,然后加入200ml丙酮,充分搅拌,使三聚氯氰分散均匀,往四口瓶中滴加94.11g(即0.5mol)的苯酚,又将20.00g(即0.5mol)的氢氧化钠溶于50ml蒸馏水并用恒压漏斗滴加到四口瓶中,控制ph在5-7,反应温度控制在10℃,反应时间为2小时,得到三聚氯氰的一元取代物;
步骤二
在另外一个装有搅拌器、恒压滴液漏斗的1000ml三口烧瓶中,加入120.20g(即2mol)的乙二胺、101.20g(即1mol)的三乙胺和200ml的三氯甲烷,冷却到-10℃,用恒压漏斗滴加97.49g(即0.5mol)的苯基磷肽二氯到三口瓶中,搅拌反应6小时,旋蒸除去未反应的化合物和三氯甲烷,得到含磷二胺中间体;
步骤三
将上述含磷二胺中间体溶解到n,n-二甲基甲酰胺中,转入恒压漏斗,缓慢滴加到第一步装有的三聚氯氰的一元取代物的四口瓶中,逐步升温到40℃,反应6小时;之后继续升温到110℃,反应10小时,反应结束;洗涤、过滤、烘干,得到浅黄色沉淀(产物2),共176.8g,产率为93.5%。本实施
例的产物结构式如下:
实施例三
步骤一
在装有回流冷凝器、温度计、搅拌器、恒压滴液漏斗的1000ml四口瓶中,加入92.25g(即0.5mol)的三聚氯氰,然后加入200ml丙酮,充分搅拌,使三聚氯氰分散均匀,往四口瓶中滴加46.56g(即0.5mol)的苯胺,将20g(即0.5mol)的氢氧化钠溶于50ml蒸馏水,用恒压漏斗滴加,控制ph在5-7,反应温度控制在0℃,反应时间为1小时,得到三聚氯氰的一元取代物;
步骤二
在另外一个装有搅拌器、恒压滴液漏斗的1000ml三口烧瓶中,加入120.20g(即2mol)的乙二胺、101.20g(即1mol)的三乙胺和200ml的三氯甲烷,冷却到-5℃,用恒压漏斗滴加89.50g(即0.5mol)的苯基二氯化磷到三口瓶中,搅拌反应3小时,旋蒸除去未反应的化合物和二氯甲烷,得到含磷二胺中间体;
步骤三
将上述含磷二胺中间体溶解到二甲亚砜中,转入恒压漏斗,缓慢滴加到第一步装有三聚氯氰的一元取代物的四口瓶中,逐步升温到45℃,反应5小时;之后继续升温到150℃,反应5小时,反应结束;洗涤、过滤、烘干,得到浅黄色沉淀(产物3),共170.8g,产率为94.1%。本实施例产物的结
构式如下:
以上结合实施例对本发明作出详细说明,但本发明不局限于所描述的实施方式。对于本领域的普通技术人员而言,在不脱离本发明的原理和宗旨的情况下对这些实施方式进行多种变化、修改、替换及变形仍落入在本发明的保护范围内。
- 还没有人留言评论。精彩留言会获得点赞!