一种可交联超支化聚酰亚胺及其制备方法与应用与流程
本发明属于交联超支化聚酰亚胺制备技术领域。
背景技术:
作为特种工程塑料的聚酰亚胺具有较好的耐热和耐低温性能、突出的机械性能、卓越的尺寸稳定性、良好的化学稳定性等优异的综合性能,已广泛应用于航天航空、气体吸附材料、微电子器件、涂料等相关领域。此外超支化聚酰亚胺由于其多维度的空间网状结构更赋予了超支化聚酰亚胺在气体吸附等领域的应用。
自交联聚酰亚胺不需要借助外加交联剂,在一定引发条件下聚合物自身的活性基团之间发生反应形成交联结构。与外加交联剂相比,自交联可控性更强。根据引发方式不同,可分为紫外引发和热引发自交联两种。其中热引发自交联形成的结构具有更高的热稳定性,作为热引发自交联的活性基团苯乙炔基交联时有着不释放副产物,无需催化剂等特点,因此得到学者们的广泛研究。
关绍巍,姚洪岩等人于2014年申请的名称为一种含苯乙炔基二酐单体及其合成方法和应用的发明专利(cn104478838a),以及在2017年发表的sci文章“microporouspolyimidesnetworksconstructedthroughatwo-steppolymerizationapproach,andtheircarbondioxideadsorptionperformance”(polym.chem.,2017,8,1298-1305)和“fromaflexiblehyperbranchedpolyimidetoamicroporouspolyimidenetwork:microporousarchitectureandcarbondioxideadsorption”(polymer,2017,115:176-183);但是通过该方法合成出的聚合物由于二酐单体中引入的苯乙炔基在联苯基团上,导致了交联侧链变长,进而导致合成的聚合物孔径大,不利于气体吸附的问题。
技术实现要素:
为了解决上述问题,本发明设计合成了一种可交联超支化聚酰亚胺,利用了苯乙炔基具有热引发交联不会产生副产物,且无需催化剂,交联后刚性大等特点,将苯乙炔基引入到聚合物体系中,同时缩短交联侧链长度,这样经过热交联处理得到交联的聚酰亚胺材料,提高了聚合物热性能,机械性能,并且生成网络结构,相比于上述现有技术还具有孔径分布小,微孔体积更大的,气体吸附效果更好的特点,进一步扩展了交联聚酰亚胺在气体吸附材料领域的应用。
所述的可交联超支化聚酰亚胺,在以酸酐封端时,结构式为
在以氨基封端时,结构式为
中的任意一种。
所述的可交联超支化聚酰亚胺的制备步骤如下:
a、二酐单体制备,合成路线如下:
步骤1):将摩尔比为1:(2~2.4)的2-溴-1,4-对苯二酚与4-硝基邻苯二甲腈投入到三口瓶中,2-溴-1,4-对苯二酚首先与摩尔的量为其2~2.4倍的碳酸铯成盐,之后在反应溶剂中与4-硝基邻苯二甲腈室温反应12~48小时,其中反应固含量约为20%~40%,反应结束后出料于去离子水中,并洗涤3~4次至滤液澄清,得到的固体粉末烘干后用乙腈和水的混合溶液重结晶得到含溴的四腈单体;
步骤2):将步骤1)的产物含溴的四腈单体与三苯基膦投入三口瓶中,加入反应溶剂,常温搅拌至固体溶解后加入与反应物摩尔比为1:(1.2~1.5)的苯乙炔,双三苯基膦二氯化钯,和三乙胺,提高反应温度至60℃,加入碘化亚铜和三乙胺,提高反应温度至80℃且反应8~24小时,蒸出部分三乙胺后出于稀盐酸中过滤并用去离子水洗涤3~4次至滤液澄清,得到黄色粉末,并用乙腈重结晶得到侧基含苯乙炔基的四腈单体;
步骤3):将摩尔比为1:(20~40)的步骤2)的产物与氢氧化钾投入三口瓶中,在乙醇和水的混合溶剂中加热回流12~80小时,反应结束后冷却至室温,加入稀盐酸调节ph值为2~3,过滤得到的固体用去离子水洗涤3~4次至滤液澄清,真空80℃烘24小时,得到含有四羧基的水解产物;
步骤4):将步骤3)的产物加入到乙酸和乙酸酐的混合溶剂中加热回流6~10小时,趁热过滤,将冷却得到的固体真空100℃烘24~48小时,得到最终产物4,4,-(苯乙炔基对苯)二醚二酐单体。
步骤1)和步骤2)中所述的反应溶剂为n,n-二甲基甲酰胺,n,n-二甲基乙酰胺或n-甲基吡咯烷酮;在步骤2)中,三苯基膦,双三苯基膦二氯化钯和碘化亚铜的摩尔总量与含溴的四腈单体的摩尔比为(0.01~0.1):1;
在步骤3)中,所述的固含量为反应物质量与溶剂体积的比值;
在步骤2)中,n,n-二甲基甲酰胺或n,n-二甲基乙酰胺和三乙胺的溶剂体积比为(1~10):(1~10);在步骤3)中,所述的乙醇和水的混合溶剂体积比为1:1;在步骤4)中,所述的乙酸和乙酸酐的混合溶剂体积比为(1~3):(1~3)。
b、超支化聚酰亚胺的制备,
b1、酸酐封端的超支化聚酰亚胺的制备步骤如下:
侧基含苯乙炔基的二醚二酐单体溶解于n-甲基吡咯烷酮中得到二醚二酐溶液,将溶解在n-甲基吡咯烷酮中的芳香多元胺溶液逐滴加入到二醚二酐溶液中,室温反应8~16小时,向反应体系中加入催化剂量的异喹啉,升温至120℃反应5~6小时,反应体系升温到180℃反应16~24小时,降温出料于无水乙醇中过滤,用索氏提取器以乙醇为溶剂洗涤48小时,真空100℃烘48~60小时得到酸酐封端的超支化聚酰亚胺;其中侧基含苯乙炔基的二醚二酐单体与芳香多元胺的摩尔比为(2~3):1;
b2、氨基封端的超支化聚酰亚胺的制备步骤如下:
侧基含苯乙炔基的二醚二酐单体溶解于n-甲基吡咯烷酮中得到二醚二酐溶液,将二醚二酐溶液逐滴加入到溶解在n-甲基吡咯烷酮中的芳香多元胺溶液中,室温反应8~16小时,向反应体系中加入催化剂量的异喹啉,升温至120℃反应5~6小时,反应体系升温到180℃反应16~24小时,降温出料于无水乙醇中过滤,用索氏提取器以乙醇为溶剂洗涤48小时,真空100℃烘48~60小时得到氨基封端的超支化聚酰亚胺;其中侧基含苯乙炔基的二醚二酐单体与芳香多元胺的摩尔比为1:(1~2);
其中所述的芳香多元胺是三聚氰胺、三(4-氨基苯基)胺、1,3,5-三(4-氨基苯基)苯、1,3,5-三(4-氨基苯氧基)苯、1,3,5-三(4-氨基-2-2三氟甲基苯氧基)苯、2,4,6-三(4-氨基苯基)吡啶、2,4,6-三-(4-氨基苯氧基)-1,3,5-三嗪、三(3-氨基苯基)氧化三苯磷、2,2’,7,7’-四氨基-9,9’-螺二芴、四(4-氨基苯基)甲烷、4,4’,4”,4”’-四氨基酞菁或铜、锌、铁、钴、镍的4,4’,4”,4”’-四氨基酞菁螯合物、4-四氨基苯基卟啉或铜、锌、铁、钴、镍的4,4’,4”,4”’-四氨基苯基卟啉螯合物。
上述的可交联聚酰亚胺在经过热交联处理后得到交联聚酰亚胺,热交联处理方法如下:
将b1或b2步骤所得的超支化聚酰亚胺转移至容器中,以二苯砜为反应溶剂,加热到360℃,高温交联反应24~36小时,降温至150℃时出于丙酮中,所得到的固体用丙酮作为溶剂用索氏提取器洗涤48小时,真空100~120℃烘36~48小时得到交联超支化聚酰亚胺。
本发明的有益效果:
本发明提供的交联超支化聚酰亚胺,交联密度高且可控度好。聚合物热性能,机械性能,并且生成网络结构,相比于cn104478838a所公开的方案还具有孔径分布小,微孔体积更大、气体吸附效果更好的特点,进一步扩展了交联聚酰亚胺在气体吸附材料领域的应用。
附图说明
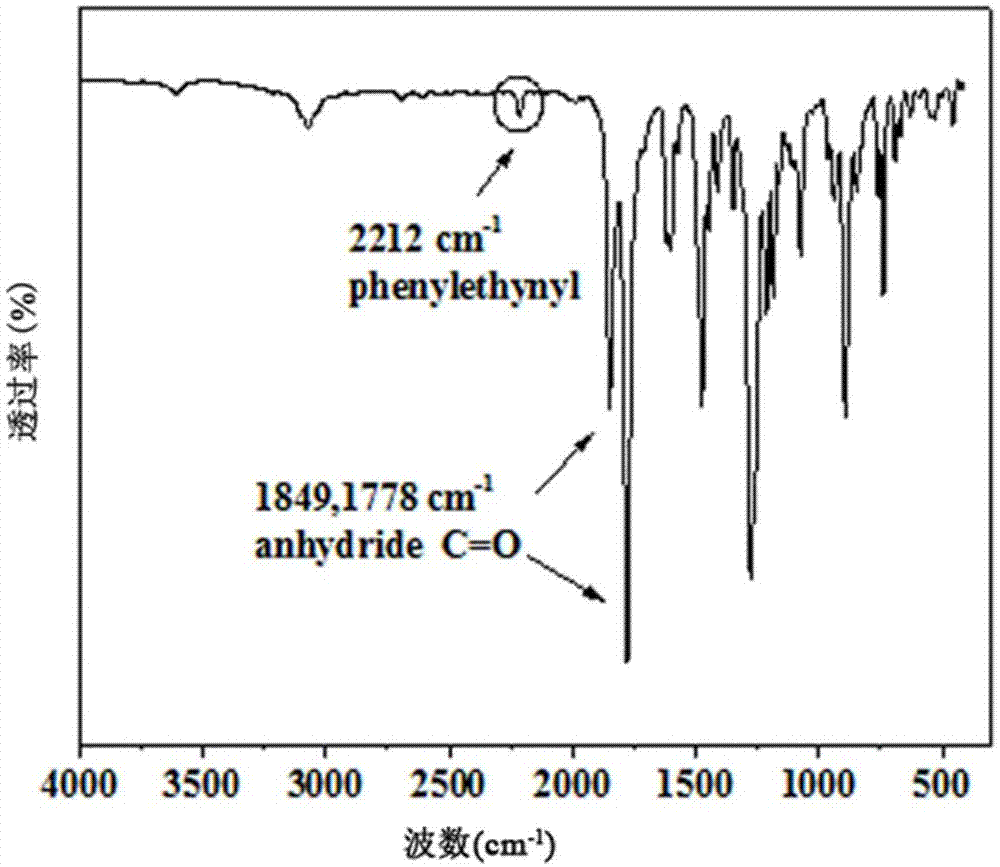
图1是4,4,-(苯乙炔基对苯)二醚二酐的红外光谱图;
图2是4,4,-(苯乙炔基对苯)二醚二酐的氢核磁谱图;
图3是4,4,-(苯乙炔基对苯)二醚二酐的碳核磁谱图;
图4是以4,4,-(苯乙炔基对苯)二醚二酐为二酐,四(4-氨基苯基)甲烷为四胺制备的酸酐封端的未交联超支化聚酰亚胺和交联超支化聚酰亚胺的红外光谱图;
图5是以4,4,-(苯乙炔基对苯)二醚二酐为二酐,四(4-氨基苯基)甲烷为四胺制备的酸酐封端的未交联超支化聚酰亚胺和交联超支化聚酰亚胺的tga曲线;
图6是以4,4,-(苯乙炔基对苯)二醚二酐为二酐,四(4-氨基苯基)甲烷为四胺制备的酸酐封端的未交联超支化聚酰亚胺(a)和交联超支化聚酰亚胺(b)的77k氮气吸附曲线;
图7是以4,4,-(苯乙炔基对苯)二醚二酐为二酐,四(4-氨基苯基)甲烷为四胺制备的酸酐封端的未交联超支化聚酰亚胺和交联超支化聚酰亚胺的77k氮气吸附的孔径分布曲线;
图8是以4,4,-(苯乙炔基对苯)二醚二酐为二酐,四(4-氨基苯基)甲烷为四胺制备的酸酐封端的未交联超支化聚酰亚胺(a)和交联超支化聚酰亚胺(b)的扫描电镜图像。
具体实施方式
下面以具体实施例的形式对本发明技术方案做进行进一步解释和说明。
实施例1:4,4,-(苯乙炔基对苯)二醚二酐的制备
4,4,-(苯乙炔基对苯)二醚二酐的制备方法如下:
步骤(1):将摩尔比为1:(2~2.4)的2-溴-1,4-对苯二酚与4-硝基邻苯二甲腈投入到三口瓶中,2-溴-1,4-对苯二酚首先与摩尔的量为其2~2.4倍的碳酸铯成盐,之后在反应溶剂中与4-硝基邻苯二甲腈室温反应12~48小时,其中反应固含量约为20%~40%,反应结束后出料于去离子水中,并洗涤3~4次至滤液澄清,得到的固体粉末烘干后用乙腈和水的混合溶液重结晶得到含溴的四腈单体;
步骤(2):将步骤(1)产物含溴的四腈单体与三苯基膦投入三口瓶中,加入反应溶剂,常温搅拌至固体溶解后加入与反应物摩尔比为1:(1.2~1.5)的苯乙炔,双三苯基膦二氯化钯,和三乙胺,提高反应温度至60℃,加入碘化亚铜和三乙胺,提高反应温度至80℃且反应8~24小时,蒸出部分三乙胺后出于稀盐酸中过滤并用去离子水洗涤3~4次至滤液澄清,得到黄色粉末,并用乙腈重结晶得到侧基含苯乙炔基的四腈单体;
步骤(3):将摩尔比为1:20~40的步骤(2)产物与氢氧化钾投入三口瓶中,在乙醇和水的混合溶剂中加热回流12~80小时,反应结束后冷却至室温,加入稀盐酸调节ph值为2~3,过滤得到的固体用去离子水洗涤3~4次至滤液澄清,真空80℃烘24小时,得到含有四羧基的水解产物;
步骤(4):将步骤(3)产物加入到乙酸和乙酸酐的混合溶剂中加热回流6~10小时,趁热过滤,将冷却得到的固体真空100℃烘24~48小时,得到最终产物4,4,-(苯乙炔基对苯)二醚二酐单体。
反应溶剂为n,n-二甲基甲酰胺,n,n-二甲基乙酰胺或n-甲基吡咯烷酮;在反应(2)中,三苯基膦,双三苯基膦二氯化钯和碘化亚铜的摩尔总量与含溴的四腈单体的摩尔比为(0.01~0.1):1。
在反应(1)中,所述的固含量为反应物质量与溶剂体积的比值。
在反应(2)中,n,n-二甲基甲酰胺或n,n-二甲基乙酰胺和三乙胺的溶剂体积比为1~10:1~10;在反应(3)中,所述的乙醇和水的混合溶剂体积比为1:1;在反应(4)中,所述的乙酸和乙酸酐的混合溶剂体积比为(1~3):(1~3)。
图1给出的是4,4,-(苯乙炔基对苯)二醚二酐的红外光谱图,1849和1788cm-1处分别对应4,4,-(苯乙炔基对苯)二醚二酐羰基的非对称和对称的伸缩振动,2212cm-1处对应碳碳三键的特征吸收峰;
图2和图3给出的是4,4,-(苯乙炔基对苯)二醚二酐的氢核磁谱图和碳核磁氢谱图,图中标注的化学位移与积分面积分别对应于4,4,-(苯乙炔基对苯)二醚二酐的结构,证明已制备出了4,4,-(苯乙炔基对苯)二醚二酐单体;
实施例2:以四(4-氨基苯基)甲烷为四胺单体,4,4,-(苯乙炔基对苯)二醚二酐为二酐,酸酐封端的超支化聚酰亚胺的制备方法如下:
侧基含苯乙炔基的二醚二酐单体溶解于n-甲基吡咯烷酮中得到二醚二酐溶液,将溶解在n-甲基吡咯烷酮中的四(4-氨基苯基)甲烷溶液逐滴加入到二醚二酐溶液中,室温反应8~16小时,向反应体系中加入催化剂量的异喹啉,升温至120℃反应5~6小时,反应体系升温到180℃反应16~24小时,降温出料于无水乙醇中过滤,用索氏提取器以乙醇为溶剂洗涤48小时,真空100℃烘48~60小时得到酸酐封端的超支化聚酰亚胺;其中侧基含苯乙炔基的二醚二酐单体与四(4-氨基苯基)甲烷的摩尔比为(2~3):1;
将上述得到的4,4,-(苯乙炔基对苯)二醚二酐封端的超支化聚酰亚胺投入三口瓶,以二苯砜为反应溶剂,加热到360℃,高温交联反应24~36小时,降温至150℃时出于丙酮中,所得到的固体用丙酮作为溶剂用索氏提取器洗涤48小时,真空100~120℃烘36~48小时得到交联超支化聚酰亚胺。
4,4,-(苯乙炔基对苯)二醚二酐封端的超支化聚酰亚胺及交联超支化聚酰亚胺的制备路线如下:
图4给出的是以4,4,-(苯乙炔基对苯)二醚二酐为二酐,四(4-氨基苯基)甲烷为四胺制备的酸酐封端的未交联超支化聚酰亚胺和交联超支化聚酰亚胺的红外光谱图,所有聚合物的红外曲线中都没有1650cm-1酰胺羰基的吸收峰,说明亚胺化进行完全;在热交联处理之后,位于2211cm-1的苯乙炔基的吸收峰完全消失,表明超支化聚酰亚胺侧链的苯乙炔基之间发生交联反应,成功地制备了交联超支化聚酰亚胺。
图5是以4,4,-(苯乙炔基对苯)二醚二酐为二酐,四(4-氨基苯基)甲烷为四胺制备的酸酐封端的未交联超支化聚酰亚胺和交联超支化聚酰亚胺的tga曲线,图中的交联超支化聚酰亚胺在升温后的初始阶段(<200℃)就出现了失重,主要是由于多孔网络中吸附的溶剂和气体造成的;交联超支化聚酰亚胺的主链分解发生在500℃以后,并且在800℃时的残炭率超过65%,说明热交联后聚合物具有较好的热稳定性。
图6是以4,4,-(苯乙炔基对苯)二醚二酐为二酐,四(4-氨基苯基)甲烷为四胺制备的酸酐封端的未交联超支化聚酰亚胺(a)和交联超支化聚酰亚胺(b)的77k氮气吸附曲线,未交联的超支化聚酰亚胺经计算bet表面积为122m2g-1,总孔体积为0.58m3g-1,微孔体积为0.007m3g-1;经过热交联处理之后,交联超支化聚酰亚胺表现出典型微孔材料的吸附曲线,经计算交联超支化聚酰亚胺的bet表面积342m2g-1,总孔体积为0.15m3g-1,微孔体积为0.10m3g-1,微孔体积明显上升,表明成功制备了多孔吸附的超支化聚酰亚胺。
图7是以4,4,-(苯乙炔基对苯)二醚二酐为二酐,四(4-氨基苯基)甲烷为四胺制备的酸酐封端的未交联超支化聚酰亚胺和交联超支化聚酰亚胺的77k氮气吸附的孔径分布曲线,采用非局部密度泛函理论计算得到了超支化聚酰亚胺的孔径分布,热交联处理前聚合物中有大量的介孔和大孔,同时具有少量的微孔;360℃热交联处理之后,使得聚合物结构中存在着大量的微孔并且具有较窄的孔径分布,主要集中在1.27nm附近。
图8是以4,4,-(苯乙炔基对苯)二醚二酐为二酐,四(4-氨基苯基)甲烷为四胺制备的酸酐封端的未交联超支化聚酰亚胺(a)和交联超支化聚酰亚胺(b)的扫描电镜图像,从图中可以看出热交联处理后(b)相比于处理前(a)展现出更高度聚集的纳米聚合物粒子堆积形成的结构,利于形成稳定的微孔结构,这是该种材料能够用于气体吸附材料领域的主要原因。
实施例3:以4,4,-(苯乙炔基对苯)二醚二酐为二酐,四(4-氨基苯基)甲烷为四胺单体,氨基封端的超支化聚酰亚胺的制备方法如下:
侧基含苯乙炔基的二醚二酐单体溶解于n-甲基吡咯烷酮中得到二醚二酐溶液,将二醚二酐溶液逐滴加入到溶解在n-甲基吡咯烷酮中的四(4-氨基苯基)甲烷溶液中,室温反应8~16小时,向反应体系中加入催化剂量的异喹啉,升温至120℃反应5~6小时,反应体系升温到180℃反应16~24小时,降温出料于无水乙醇中过滤,用索氏提取器以乙醇为溶剂洗涤48小时,真空100℃烘48~60小时得到氨基封端的超支化聚酰亚胺;其中侧基含苯乙炔基的二醚二酐单体与四(4-氨基苯基)甲烷的摩尔比为1:(1~2)。
实施例4:以三聚氰胺为三胺单体代替四(4-氨基苯基)甲烷,重复实施例2和3,制备酸酐封端的和氨基封端的超支化聚酰亚胺。
实施例5:以1,3,5-三(4-氨基苯基)苯为三胺单体代替四(4-氨基苯基)甲烷,重复实施例2和3,制备酸酐封端的和氨基封端的超支化聚酰亚胺。
实施例6:以四氨基苯基卟啉为四胺单体,制备酸酐封端的超支化聚酰亚胺的具体制备方法如下:
侧基含苯乙炔基的二醚二酐单体溶解于n-甲基吡咯烷酮中得到二醚二酐溶液,将溶解在n-甲基吡咯烷酮中的四氨基苯基卟啉溶液逐滴加入到二醚二酐溶液中,室温反应8~16小时,向反应体系中加入催化剂量的异喹啉,升温至120℃反应5~6小时,反应体系升温到180℃反应16~24小时,降温出料于无水乙醇中过滤,用索氏提取器以乙醇为溶剂洗涤48小时,真空100℃烘48~60小时得到酸酐封端的超支化聚酰亚胺;其中侧基含苯乙炔基的二醚二酐单体与四氨基苯基卟啉的摩尔比为(2~3):1;
实施例7:以三(4-氨基苯基)胺为三胺单体代替四(4-氨基苯基)甲烷,重复实施例6,制备酸酐封端的和氨基封端的超支化聚酰亚胺。
实施例8:以1,3,5-三(4-氨基苯氧基)苯为三胺单体代替四(4-氨基苯基)甲烷,重复实施例6,制备酸酐封端的和氨基封端的超支化聚酰亚胺。
- 还没有人留言评论。精彩留言会获得点赞!