Nfdirp抗旱基因、其编码的氨基酸序列及其在提高植物抗旱性中的用途的制作方法
本发明属于基因工程领域,特别涉及一种来源于发状念珠藻的nfdirp抗旱基因及其编码的氨基酸序列,用于提高植物特别是水稻的抗旱性。
背景技术:
发状念珠藻(nostocflagelliforme)主要分布在北半球的干旱或半干旱的荒漠地区,经常受到极度干旱、较大温差、高浓度盐碱、营养缺乏、uv-b辐射等非生物因素的胁迫。胁迫因子可能会对细胞产生一系列损伤,包括核酸损伤、蛋白质损伤、膜脂损伤等,从而扰乱正常的细胞代谢。经过长期的环境选择和自身结构功能的适应,发状念珠藻已经进化出一系列的生理生态和分子的机制来抵抗各种逆境。因此,发状念珠藻已经成为研究逆境适应机制和发掘抗逆基因的最佳材料之一。
rna结合蛋白(rbps)在基因调控过程中扮演着关键作用,目前除了少数rna能以核酶的形式单独发挥功能,大部分rna是与蛋白结合形成rna-蛋白复合物,rbps对于rna的合成、选择性剪接、修饰、转运和翻译等生命活动的调控都具有关键作用。研究表明,rna结合蛋白参与了植物对多种逆境条件的反应调节,如冷害、干旱、水涝、高盐、aba、sa等。
我国是个自然灾害频发的国家,水资源相对贫乏,在地理和时空上分布极不均匀,使我国的农业经常遭受干旱而造成粮食减产。水稻是我国最重要的粮食作物之一,其生产消耗掉我国近一半的淡水资源,提高水稻等农作物的节水抗旱性能,是保障我国粮食安全和生态安全,保障农业可持续发展的重大需求(luo,2010),目前转基因技术已被广泛应用到抗旱水稻的培育中,所采用的策略是在水稻中表达干旱诱导或抗旱相关基因。因此,对发状念珠藻抗旱基因的克隆和挖掘,并应用在生物基因工程上具有非常重要意义。
技术实现要素:
鉴于此,本发明的目的是提供一种来源于发状念珠藻的nfdirp抗旱基因,基于发状念珠藻干旱胁迫转录得到,其在干旱胁迫下表达量明显上升(图1),命名为nfdirp抗旱基因(drought-inducedrnabindingprotein)。
本发明的再一目的是提供上述nfdirp抗旱基因在提高植物抗旱性中的用途,尤其是在水稻中的用途。
本发明的上述目的通过以下技术方案实现:
第一方面,nfdirp抗旱基因,来源于发状念珠藻,其核苷酸序列如seqidno.1所示,序列长度为300bp,具体为:
atgtcagtttatgtaggcaatctttcttacgaagttacagaagaaggcttgaattctgtgtttgcagaatatggttctgtaaagcgggttcagctacctaccgaccgtgagacaggtcgtatgcgtggctttggttttgtggaaatgggcacagatgctgaagaaacagctgccattgaagctcttgatggtgctgagtggatgggacgtgacctaaaagtgaacaaggctaaacccagagaagacagaggttcctttggtggaaatcggggaaacaatagtttcggcaatcgttactaa;其编码的氨基酸序列为99个,如seqidno.2所示,具体为:
msvyvgnlsyevteeglnsvfaeygsvkrvqlptdretgrmrgfgfvemgtdaeetaaiealdgaewmgrdlkvnkakpredrgsfggnrgnnsfgnry。
第二方面,上述nfdirp抗旱基因在提高植物抗旱性上的用途,所述植物为水稻。
根据所述nfdirp抗旱基因序列,直接采用pcr(polymerasechainreaction)技术,从基因组、mrna和cdna中扩增得到所述nfdirp抗旱基因以及任何感兴趣的一段dna或与其同源的一段dna,携带上述nfdirp抗旱基因的表达载体可通过使用ti质粒,植物病毒载体,直接dna转化,微注射,电穿孔等常规生物技术方法导入原核细菌细胞,植物细胞中。通过分离和应用发状念珠藻中包含nfdirp抗旱基因的dna片段,可以使大肠杆菌和水稻在干旱条件下的抗旱能力增强。
本发明的有益效果在于:
将野生型种子和转nfdirp基因水稻t2种子进行发芽试验,经过在添加120mm甘露醇的1/2ms培养基培养10天后,野生型平均株高为6.15cm的小芽,而转基因株系长势明显优于野生型对照,转基因家系平均株高均超过了10cm,证实在水稻中超表达nfdirp抗旱基因提高了转基因植株的抗旱性。
附图说明
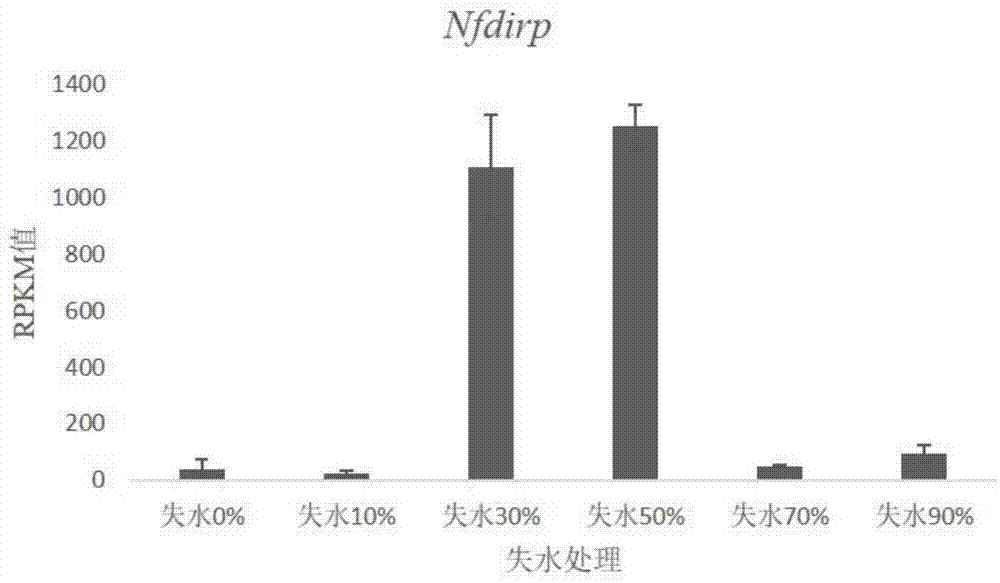
图1是实施例1中nfdirp抗旱基因在发菜失水转录组的表达情况对比图。
图2是实施例1中nfdirp抗旱基因在失水胁迫下的表达分析。
图3是实施例2中nfdirp抗旱基因的原核表达sds-page电泳结果图,其中,ck1、ck2为空载体阴性对照,1,2,3,4,5,6为6个重组质粒pgex-nfdirp克隆。
图4是实施例2中原核表达胁迫结果;pgex为空载体,nfdirp为重组质粒。其中,图4a为正常培养对照条件下实验结果图,图4b为添加山梨醇条件下实验结果图,图4c为同时添加iptg和山梨醇的实验结果图。
图5是本发明选用的植物超量表达载体ub-06载体图。
图6是实施例3中转nfdirp抗旱基因水稻株系的表达量检测图。
图7是实施例4中的转nfdirp抗旱基因水稻株系oe-1、oe-2、oe-4与野生型wt在添加120mm甘露醇的平板中生长10天后的株高统计(7a);oe-1、oe-2、oe-4和野生型wt在添加120mm甘露醇的平板中生长10天后的生长表现(7b)。
具体实施方式
下面结合实施例,对本发明作进一步说明。
下述实施例中未注明具体条件的实验方法,通常按照常规条件,例如sambrook等分子克隆:实验室手册(newyork:coldspringharborlaboratorypress,1989)中所述的条件,或按照制造厂商所建议的条件。
实施例1:nfdirp抗旱基因的克隆
(1)发状念珠藻的培养
液体培养的发状念珠藻(天然野生样品采样于内蒙古自治区苏尼特左旗)。接种前用玻璃匀浆器匀浆藻种使藻丝分散,然后接种到300ml,bg11培养基中进行无菌培养,培养温度25℃,光强20±2μmolphotonsm-2s-1(24h连续光照),每天早中晚摇藻三次,使藻细胞悬浮、均匀接受光照。
(2)dna提取
将步骤(1)中培养的发状念珠藻样品在液氮中研磨,分装到1.5ml离心管中,加入1ml提取液(100mmol/l的tris-hcl,500mmol/l的nacl,50mmol/l的edta,ph8.0),100微升20%sds,10微升10mg/ml蛋白酶k,充分混匀;37℃裂解30min,每5min混匀一次;12000rpm离心10min,取上清加入等体积tris饱和酚,充分混匀,室温静置2min;12000rpm离心10min,取上清加入等体积氯仿/异戊醇,充分混匀,室温静置2min;12000rpm离心10min,取上清加入等体积氯仿,充分混匀,室温静置2min;12000rpm离心10min,取上清加入等体积异丙醇,加入1/10体积的3mnaac,充分混匀,可见白色丝状物;用10微升吸头挑取白色丝状沉淀,转入70%乙醇洗2次;空气中晾干,加入30微升buffereb溶解。
(3)nfdirp全长序列的扩增
在发状念珠藻失水转录组分析中,在失水10%,失水30%,失水50,失水70%,失水90%的条件下,nfdirp抗旱基因的表达都显著升。通过转录组测序获得nfdirp抗旱基因的全长序列,根据序列信息设计上下游引物(nfdirpf1:5-atgtcagtttatgtaggcaa-3,nfdirpr1:5-ttagtaacgattgccgaaac-3),如序列表seqidno.3-4所示。
从发状念珠藻基因组dna中克隆获得了nfdirp抗旱基因,通过测序验证,扩增产物是如seqidno.1所示的核苷酸序列(1-300bp)。
(4)nfdirp抗旱基因在失水处理下的表达谱分析
将固体发菜浸泡在bg11培养基中,2天后达到完全饱和。并开始失水处理,在失水0%,失水10%,失水30%,失水50%,失水70%和失水90%6个时间点取样,置液氮中研磨。使用tiangen公司的rnapreppure多酚多糖植物总rna提取试剂盒(目录号:dp441)提取发菜的总rna,电泳鉴定总rna质量,然后在分光光度计上测定rna含量。
逆转录合成第一链cdna,反转录之前需用dnasei消化所提取rna样品,反应体系如下:
37℃反应15min后,加入0.25μl0.1medta(保证终浓度>2mm),70℃温育10min终止反应,短暂离心后置于冰上备用。
第一链cdna的合成参照promega反转录系统a3500操作手册,反应体系如下:
将上反应体系于42℃温育15min;然后95℃加热5min,使amv反转录酶失活并阻止其与dna结合;4℃或冰上放置5min。制备好的cdna可以立即使用或存放于-20℃备用。
基因表达的定量分析使用takara公司的
反应条件为:95℃30s,然后在95℃5s,60℃31s,循环40次,并增设dissociationstage。设定在每个循环中60℃31s时收集数据,其它具体操作按仪器使用说明书进行。计算目的基因与参照基因的平均ct值以及△ct值,利用2-δδct法进行结果分析,最后将数据导入graphpadprism5.0做出目的基因的相对表达量柱状图,见图2。
实施例2:发状念珠藻nfdirp蛋白的原核表达及胁迫鉴定
利用onestepcloningkit重组技术构建nfdirp的原核表达载体。以实施例1中已获得nfdirp抗旱基因为模板,用前引物nfdirpf2:5-ttccaggggcccctgggatccatgtcagtttatgtaggcaa-3,后引物nfdirpr2:5-gtcacgatgcggccgctcgagttagtaacgattgccgaaac-3,引物如序列表seqidno.7-8所示,进行pcr扩增,产物经回收纯化后,通过onestepcloningkit重组反应,将扩增产物片段克隆到载体pgex-6p-14984上。具体过程如下:
(1)pcr扩增
反应体系如下:
扩增程序:94℃预变性3min,94℃1min,56℃45sec,72℃1min,30个循环,72℃10min。反应结束后取5μl电泳检测。
(2)pcr产物的回收
采用普通琼脂糖凝胶dna回收试剂盒(天根生化科技有限公司)进行纯化回收。
(3)重组反应
采用南京诺唯赞生物科技有限公司的onestepcloningkit试剂盒进行重组反应。具体如下:
将2μg环状pgex-6p-14984质粒加入到20μl酶切反应体系中,37℃酶切2hr。限制性内切酶使用量为bamhi和xhoi各1μl。酶切完成后,将酶切产物置于65℃加热20min,失活内切酶。
于冰水浴中配制如下反应体系:
体系配制完成后,用移液器上下轻轻吹打几次混匀各组分,置于37℃反应30min。待反应完成后,立即将反应管置于冰水浴中冷却5min。然后取20μl冷却反应液,加入到200μl表达感受态bl21细胞中,轻弹管壁数下混匀,在冰上放置30min。42℃热激45~90秒,冰水浴孵育2min。加入900μllb培养基,37℃孵育10min充分复苏。37℃摇菌45min。取100μl菌液均匀涂布在含有抗生素amp+的平板上。将平板倒置,于37℃过夜培养,测序获得含nfdirp的重组质粒pgex-nfdirp。
(4)nfdirp蛋白的表达
将含nfdirp重组质粒的菌株单克隆置于2mllb(含amp50μg/ml)中37℃过夜培养。按1∶50比例稀释过夜菌,37℃震荡培养至od600≌0.6,取部分液体作为未诱导的对照组,余下的加入iptg诱导剂至终浓度0.4mm作为实验组,两组继续37℃震荡培养3hr。分别取菌体1ml,离心12000g×30s收获沉淀,用100μl1%sds重悬,混匀,70℃10min。离心12000g×1min,取上清作为样品,sds-page电泳检测,具体结果如图3所示。其中,ck1和ck2为空载体阴性对照,1,2,3,4,5,6为6个重组质粒pgex-nfdirp克隆。
(5)原核表达胁迫
按步骤(4)所述,摇菌待od600处于0.4至0.6之间,pgex空载体作为对照,调节各管的初始od600一致,然后取同等量扩繁的菌液加入各种处理的含氨苄lb中,3种处理分别为不加任何额外的试剂、加入山梨醇(sorbitol)、加入诱导剂(iptg)与山梨醇试剂。每隔一段时间用分光光度计测定od600(每隔1h测定),统计结果绘制折线图,结果显示在未加iptg诱导条件下,重组质粒和空载体对照菌长势没有差别,而诱导后,重组质粒pgex-nfdirp在山梨醇胁迫下长势更好,具体结果如图4所示,其中图4a为正常培养对照条件下实验结果图,图4b为添加山梨醇条件下实验结果图,图4c为同时添加iptg和山梨醇的实验结果图。
实施例3:发状念珠藻nfdirp超量表达转化水稻
以实施例1中已获得nfdirp抗旱基因为模板,用前引物nfdirpf3:5-caggtcgactctagaggatccatgtcagtttatgtaggcaa-3,后引物nfdirpr3:5-gggaaattcgagctggtcaccttagtaacgattgccgaaac-3,引物如序列表seqidno.9-10所示,进行pcr扩增,产物经回收纯化后,通过onestepcloningkit重组反应,将扩增产物片段克隆至植物超量表达载体ub-06,ub-06载体图如图5所示。具体过程如下:
(1)pcr扩增,按实施案例1所述。
(2)pcr产物的回收,按实施案例2所述。
(3)重组反应
采用南京诺唯赞生物科技有限公司的onestepcloningkit试剂盒进行重组反应。将2μg环状ub-06质粒加入到20μl酶切反应体系中,37℃酶切2hr。限制性内切酶使用量为bamhi和xhoi各1μl。酶切完成后,将酶切产物置于65℃加热20min,失活内切酶。
于冰水浴中配制如下反应体系:
体系配制完成后,用移液器上下轻轻吹打几次混匀各组分,置于37℃反应30min。待反应完成后,立即将反应管置于冰水浴中冷却5min。然后取20μl冷却反应液,加入到200μl表达感受态dh5α细胞中,轻弹管壁数下混匀,在冰上放置30min。42℃热激45~90秒,冰水浴孵育2min。加入900μllb培养基,37℃孵育10min充分复苏。37℃摇菌45min。取100μl菌液均匀涂布在含有抗生素amp+的平板上。将平板倒置,于37℃过夜培养,测序获得含nfdirp的质粒u6-nfdirp。
(4)农杆菌转化
将1μlu6-nfdirp质粒加入到100μl农杆菌eha105感受态细胞中,轻轻混匀,冰水浴30min后,于液氮中速冻冷激2min;加入400-800μlyep培养液(kan+),28℃,200r/min震荡培养3-5h;室温离心(5000r/min,5min),保留100μl上清重悬菌体,涂布于la固体培养基(kan+),28℃倒置培养2天直至长出合适大小的菌落,挑取单克隆进行pcr检测,获得阳性菌株。
(5)愈伤诱导:种子用无菌水漂洗15-20min,再用75%的乙醇消毒1min,然后用次氯酸钠(1.5%有效浓度)溶液振荡消毒20min。最后再用无菌水冲洗5遍。将洗好的种子用吸水纸吸干接种在诱导愈伤培养基中,25℃暗培养2周。
愈伤诱导培养基:采用表1的诱导培养基,加入0.3g脯氨酸、0.6g水解酪蛋白酶、30g蔗糖和2.5ml2,4-d(浓度1mg/ml),配成1l溶液,调ph至5.9,加入7g琼脂粉,高温高压灭菌。
(6)继代培养:把胚性愈伤组织切下,接入继代培养基中,25℃暗培养2周。
继代培养基:采用表1的继代培养基,加入0.5g脯氨酸、0.6g水解酪蛋白酶、30g蔗糖和2ml2,4-d(浓度1mg/ml),配成1l溶液,调ph至5.9,加入7g琼脂粉,高温高压灭菌。
(7)农杆菌浸染和愈伤组织共培养:培养农杆菌,挑取阳性单菌落,在1ml农杆菌培养液(含抗生素)中,28℃培养过夜;取以上培养物,加入50ml农杆菌培养液(含抗生素)中,28℃培养至od600=0.6-1.0。将获得的农杆菌菌液离心,将收集到的菌体加入悬浮培养液中,震荡培养30min至od600=0.6-1.0。然后将愈伤组织放入含有农杆菌菌液的悬浮培养液中,振荡培养20min左右。将愈伤组织在灭菌滤纸上晾干,转入共培养培养基中,25℃暗培养5d。
悬浮培养液:采用表1的悬浮培养液,加入0.08g水解酪蛋白酶、2g蔗糖和0.2ml2,4-d(浓度1mg/ml),配成100ml溶液,调ph至5.4,分成两瓶(每瓶50ml),高温高压灭菌。使用之前加入1ml50%的葡萄糖和100μlas(100mm)。
共培养培养基:采用表1的共培养培养基,加入0.8g水解酪蛋白酶、20g蔗糖和3.0ml2,4-d(浓度1mg/ml),配成1l溶液,调ph至5.6,加入7g琼脂粉,高温高压灭菌。使用之前加入20ml50%的葡萄糖和1mlas(100mmol/l)。
(8)筛选培养:共培养3d后,选取好的愈伤组织,转入筛选培养基中,25℃暗培养2周,筛选两次。
筛选培养基:采用表2的筛选培养基,加入0.6g水解酪蛋白酶、30g蔗糖和2.5ml2,4-d(浓度1mg/ml),配成1l溶液,调ph至6.0,加入7g琼脂粉,高温高压灭菌。使用之前加入1mlhn和1mlcn(100ppm)。
(9)分化培养:挑取胚性愈伤组织接入分化培养基,24℃,16h/8h光暗培养诱导分化芽(4-6周)。
分化培养基:采用表2的分化培养基,加入2.0mg/l6-ba、2.0mg/lkt、0.2mg/lnaa、0.2mg/liaa、1.0g水解酪蛋白酶和30g蔗糖,配成1l溶液,调ph至6.0,加入7g琼脂粉,高温高压灭菌。
(10)生根培养:待芽长至2cm左右时,将幼芽切下,插入生根培养基中,25℃左右,16h/8h光暗培养,诱导生根。
生根培养基:采用表2的生根培养基,加入30g蔗糖,配成1l溶液,调ph至5.8,加入7g琼脂粉,高温高压灭菌。
(11)转化植株培养:待根系发达后,打开试管口,加入无菌水炼苗2-3d后,将植株取出,用无菌水洗净附着的固体培养基,移入土壤中,刚开始遮荫避风,待植株健壮后进行常规田间或温室管理培养。
表1:基本培养基成分1
表2:基本培养基成分2
(12)超量表达植株的阳性检测
剪取再生植株叶片,利用ctab法抽提dna,利用hpt特异引物(hptf:acactacatggcgtgatttcat;hptr:tccactatcggcgagtacttct,引物如序列表seqidno.11-12所示)进行pcr检测。
pcr体系:
pcr反应程序:
(13)超量表达阳性植株中目的基因的表达水平检测
取t0代水稻叶片,置于液氮研磨,提取rna。具体如下:
rna的提取:所取样品在研钵中用液氮冷冻后研磨成粉状,加入盛有1mltrnzol-a+试剂(天根生化科技有限公司)的2mlep管,充分振荡后,室温放置5min,之后加0.2ml氯仿,剧烈震荡15s后,室温放置3min;后于4℃,12000rpm离心10min后,上清液移至新的2mlep管中,加等体积异丙醇沉淀rna,加100μlrnase-freeddh2o溶解。电泳鉴定总rna质量,然后在分光光度计上测定rna含量。
逆转录合成第一链cdna
反转录之前需用dnasei消化所提取rna样品,反应体系如下:
37℃反应15min后,加入0.25μl0.1medta(保证终浓度>2mm),70℃温育10min终止反应,短暂离心后置于冰上备用。
第一链cdna的合成参照promega反转录系统a3500操作手册,具体步骤如下:
dnasei消化过的样品中依次加入下列各试剂配制20μl的反应体系:
将上反应体系于42℃温育15min;然后95℃加热5min,使amv反转录酶失活并阻止其与dna结合;4℃或冰上放置5min。制备好的cdna可以立即使用或存放于-20℃备用。
基因表达的定量分析使用takara公司的
反应条件为:95℃30s,然后在95℃5s,60℃31s,循环40次,并增设dissociationstage。设定在每个循环中60℃31s时收集数据,其它具体操作按仪器使用说明书进行。计算目的基因与参照基因的平均ct值以及△ct值,利用2-δδct法进行结果分析,最后将数据导入graphpadprism5.0做出目的基因的相对表达量柱状图,如图6所示。
实施例4:转基因水稻t2代种子的芽期甘露醇模拟干旱处理
将超量表达转基因家系种子去壳消毒(75%酒精处理1min,1.5%naclo处理20min,无菌水清洗5次),在含有50mg/l潮霉素的1/2ms培养基上发芽,野生型对照晚一天播于不含潮霉素的1/2ms培养基上。待发芽2~3天后挑选发芽好且长势一致的种子,转移到含120mm的1/2ms培养基中培养,10天后统计生长情况。实验结果显示经过10天胁迫培养后,野生型对照的平均株高为6.15cm,而转基因株系平均株高超过10cm,具体如图6所示,说明在水稻中超表达nfdirp抗旱基因的确提高了转基因植株的抗旱性。
以上所述为本发明的较佳实施例而已,但本发明不应该局限于该实施例所公开的内容。所以凡是不脱离本发明所公开的精神下完成的等效或修改,都落入本发明保护的范围。
序列表
<110>上海市农业生物基因中心
<120>nfdirp抗旱基因、其编码的氨基酸序列及其在提高植物抗旱性中的用途
<130>2018-10-29
<141>2018-10-31
<160>12
<170>siposequencelisting1.0
<210>1
<211>300
<212>dna
<213>未知()
<400>1
atgtcagtttatgtaggcaatctttcttacgaagttacagaagaaggcttgaattctgtg60
tttgcagaatatggttctgtaaagcgggttcagctacctaccgaccgtgagacaggtcgt120
atgcgtggctttggttttgtggaaatgggcacagatgctgaagaaacagctgccattgaa180
gctcttgatggtgctgagtggatgggacgtgacctaaaagtgaacaaggctaaacccaga240
gaagacagaggttcctttggtggaaatcggggaaacaatagtttcggcaatcgttactaa300
<210>2
<211>99
<212>prt
<213>未知()
<400>2
metservaltyrvalglyasnleusertyrgluvalthrgluglugly
151015
leuasnservalphealaglutyrglyservallysargvalglnleu
202530
prothrasparggluthrglyargmetargglypheglyphevalglu
354045
metglythraspalaglugluthralaalaileglualaleuaspgly
505560
alaglutrpmetglyargaspleulysvalasnlysalalysproarg
65707580
gluaspargglyserpheglyglyasnargglyasnasnserphegly
859095
asnargtyr
<210>3
<211>20
<212>dna
<213>未知()
<400>3
atgtcagtttatgtaggcaa20
<210>4
<211>20
<212>dna
<213>未知()
<400>4
ttagtaacgattgccgaaac20
<210>5
<211>19
<212>dna
<213>未知()
<400>5
gtatgcgtggctttggttt19
<210>6
<211>19
<212>dna
<213>未知()
<400>6
cctctgtcttctctgggtt19
<210>7
<211>41
<212>dna
<213>未知()
<400>7
ttccaggggcccctgggatccatgtcagtttatgtaggcaa41
<210>8
<211>41
<212>dna
<213>未知()
<400>8
gtcacgatgcggccgctcgagttagtaacgattgccgaaac41
<210>9
<211>41
<212>dna
<213>未知()
<400>9
caggtcgactctagaggatccatgtcagtttatgtaggcaa41
<210>10
<211>41
<212>dna
<213>未知()
<400>10
gggaaattcgagctggtcaccttagtaacgattgccgaaac41
<210>11
<211>22
<212>dna
<213>未知()
<400>11
acactacatggcgtgatttcat22
<210>12
<211>22
<212>dna
<213>未知()
<400>12
tccactatcggcgagtacttct22
- 还没有人留言评论。精彩留言会获得点赞!