一种盐酸溴己新的制备方法与流程
本发明属于有机合成技术领域,尤其涉及一种盐酸溴己新的制备方法。
背景技术:
盐酸溴己新,其化学名称为:n-甲基-n-环已基-2-氨基-3,5-二溴苯甲胺盐酸盐,药物别名:溴己铵,溴苄环己铵,必嗽平、盐酸溴已新、必消痰、必漱平、溴苄环已胺。本品为白色或类白色的结晶性粉末;无臭,无味。本品在乙醇或氯仿中略溶,在水中极微溶解。本品可直接作用于支气管腺体,促使粘液分泌细胞的溶酶体释出,使痰中的粘的糖纤维分化裂解;还可抑制粘液腺和杯状细胞中酸性糖蛋白的合成,使之分泌粘滞性较低的小分子糖蛋白,从而使痰液的粘稠度降低,易于咳出。此外,还可刺激胃粘膜反射性地引起呼吸道腺体分泌增加,使痰液稀释。支气管炎、哮喘、肺气肿等粘痰不易咳出。
目前,关于盐酸溴己新的合成工艺文献报道比较多,而综合文献分析发现,报道的盐酸溴己新的合成方法可以总结以下几种,其合成路线如下:
路线一
该条路线反应类型较为简单,硼氢化钠还原3,5-二溴-2-氨基苯甲醛为3,5-二溴-2-氨基苯甲醇,产物为黄色,纯度不够高;二氯亚砜氯代反应时,污染大,生产操作困难,而且氨基取代卤素反应会产生很多副反应产物,导致api需要多次精制才能达到质量标准,另外二氯亚砜的使用有导致基因毒性杂质的风险。
路线二
该路线(cn106631828a)是修改上述路线使用含金属试剂钛酸酯,反应完后,用水淬灭反应,产生很大量的不溶物,需要大量的有机溶剂萃取产物,导致反应后处理困难;而金属试剂当量的使用成本也较高,生成中间体判断对于车间操作要求高,工业化生产中存在反应不完全的风险,且产生大量的废水,对环境不友好。
路线三
该路线(cn104003887a)使用较便宜的原料2-硝基溴苄为起始物,经亲核取代,雷尼镍催化加氢还原硝基,最后液溴溴化、成盐得到盐酸溴己新。该路线第一步反应杂质较大,容易形成二聚物,反应不完全;催化加氢需要特殊设备;溴化反应放在最后一步合成,因溴有氧化性,会氧化叔胺产生很多氮氧化物杂质,以盐酸作为溶剂,使用过量的溴素反应,导致大量的废酸需要处理,且不原子经济。因盐酸的使用产物中发现氯代杂质,极难除去。
路线四
该路线(cn103333074b)以3,5-二溴-2-氨基苯甲醇与过量的n-甲基环己胺于乙酸催化下,在180℃下脱水反应制备到盐酸溴己新。该步骤需要高的反应温度和特殊设备,反应时间长;难以反应完全,反应结束需要高真空度减压蒸除过量的n-甲基环己胺。从而导致生产困难,收率不高。
专利(cn104610073a)以2-氨基苯甲酸甲酯为原料,盐酸为溶剂,液溴溴化得到2-氨基-3,5-二溴苯甲酸甲酯;2-氨基-3,5-二溴苯甲酸甲酯在氯化钙催化下硼氢化钾还原得到2-氨基-3,5-二溴苯甲醇;然后与n-甲基苄胺直接缩合得到溴己新,与盐酸成盐得到盐酸溴己新。发明人在重复该工艺时发现以下几个缺点:
步骤一:溴化反应,在盐酸中进行,反应难以完全,收率低,后处理困难,特别是生产苯环上的氯代杂质i:
步骤三:缩合反应,高温下苄醇与n-甲基环己胺的脱水缩合,无法进行,反应2小时,几乎无产物生成。发明人研究发现,该反应需要在酸催化下才能进行,且其成盐在乙醇中进行,盐酸与乙醇反应生产氯代乙烷为基因毒性杂质。
综上,上述或者其他一些制备路线中,存在原料或者中间体成本高,中间体不稳定,反应苛刻,环境污染大,后处理繁琐,或者所得api纯度不高。
技术实现要素:
本发明的目的在于提供一种盐酸溴己新的制备方法,本方法使用便宜、化学稳定的初始原料,操作工艺简单,杂质少,收率高,可以工业化放大生产,能够得到符合药用级别的盐酸溴己新。
为了实现上述发明目的,本发明提供以下技术方案:
本发明提供了一种盐酸溴己新的制备方法,包含如下步骤:
(1)将2-氨基苯甲酸酯类化合物、溴素、氧化剂和混合溶剂混合后进行溴代反应,生成3,5-二溴-2-氨基苯甲酸酯;
(2)将所得3,5-二溴-2-氨基苯甲酸酯、催化剂、还原剂和溶剂混合后进行还原反应,生成3,5-二溴-2-氨基苯甲醇;
(3)将所得3,5-二溴-2-氨基苯甲醇、n-甲基环己胺、混合催化剂和溶剂混合后进行缩合反应,生成溴己新;
(4)将所得溴己新和盐酸在溶剂中进行成盐反应,生成盐酸溴己新。
优选的,所述步骤(1)中2-氨基苯甲酸酯类化合物包括2-氨基苯甲酸甲酯或2-氨基苯甲酸乙酯;
所述氧化剂包括过氧化氢、间氯过氧苯甲酸和过氧乙酸中的一种或几种;
所述混合溶剂为水和有机溶剂的混合物,所述有机溶剂为四氢呋喃、乙酸乙酯和二氯甲烷中的一种或几种,所述混合溶剂中水和有机溶剂的体积比为2:(2~6)。
优选的,所述步骤(1)中2-氨基苯甲酸酯类化合物和溴素的摩尔比为1:(1~2);
所述2-氨基苯甲酸酯类化合物和氧化剂的摩尔比为1:(1~2);
所述2-氨基苯甲酸酯类化合物和混合溶剂的质量比为1:(10~15)。
优选的,所述步骤(2)中催化剂为氯化钙、氯化锂和氯化锌中的一种或几种;
所述还原剂为硼氢化钠、硼氢化锂或硼氢化钾中的一种或几种;
所述溶剂为甲醇、无水乙醇和四氢呋喃中的一种或几种。
优选的,所述步骤(2)中3,5-二溴-2-氨基苯甲酸酯、催化剂和还原剂的摩尔比为1:(1.0~1.5):(2.0~3.0);
所述3,5-二溴-2-氨基苯甲酸酯和溶剂的质量比为1:(3.5~4.5)。
优选的,所述步骤(3)中混合催化剂包含超强酸和醋酸,所述超强酸为sio2-al2o3、sio2-zro3、sio2-oso3h和sio2-tio2中的一种或几种,所述超强酸和醋酸的质量比为(0.08~0.15):(0.2~0.5);
所述溶剂为甲苯、二甲苯和环己烷中的一种或几种。
优选的,所述步骤(3)中3,5-二溴-2-氨基苯甲醇、n-甲基环己胺和混合催化剂的质量比为1:(0.9~1.0):(0.3~0.4);
所述3,5-二溴-2-氨基苯甲醇和溶剂的质量比为1:(0.01~0.2)。
优选的,所述步骤(4)中溶剂为甲醇、乙醇、异丙醇、乙酸乙酯和丙酮中的一种或几种;
所述溴己新和溶剂的质量比为1:(4~5)。
优选的,所述步骤(4)中盐酸的质量浓度为10~20%。
优选的,所述步骤(1)中溴代反应在室温下进行,溴代反应的时间为6~8h;
所述步骤(2)中还原反应的温度为35~45℃,时间为14~16h;
所述步骤(3)中缩合反应的温度为80~90℃,时间为10~14h。
本发明提供了一种盐酸溴己新的制备方法,包含如下步骤:(1)将2-氨基苯甲酸酯类化合物、溴素、氧化剂和混合溶剂混合后进行溴代反应,生成3,5-二溴-2-氨基苯甲酸酯;(2)将所得3,5-二溴-2-氨基苯甲酸酯、催化剂、还原剂和溶剂混合后进行还原反应,生成3,5-二溴-2-氨基苯甲醇;(3)将所得3,5-二溴-2-氨基苯甲醇、n-甲基环己胺、混合催化剂和溶剂混合后进行缩合反应,生成溴己新;(4)将所得溴己新和盐酸在溶剂中进行成盐反应,生成盐酸溴己新。本发明以2-氨基苯甲酸酯类化合物为原料,经溴代反应、还原反应、缩合反应和成盐反应后得到盐酸溴己新,合成路较短,成本低,中间体稳定,对环境无污染,纯度高,收率高,符合药用标准,适于工业化扩大生产。
附图说明
图1为2-氨基苯甲酸甲酯和溴素反应的原理示意图;
图2为盐酸溴己新的uplc谱图;
图3为盐酸溴己新和氯代杂质的uplc对比图;
图4为盐酸溴己新的1hnmr谱图;
图5为盐酸溴己新的13cnmr谱图。
具体实施方式
本发明提供了一种盐酸溴己新的制备方法,包含如下步骤:
(1)将2-氨基苯甲酸酯类化合物、溴素、氧化剂和混合溶剂混合后进行溴代反应,生成3,5-二溴-2-氨基苯甲酸酯;
(2)将所得3,5-二溴-2-氨基苯甲酸酯、催化剂、还原剂和溶剂混合后进行还原反应,生成3,5-二溴-2-氨基苯甲醇;
(3)将所得3,5-二溴-2-氨基苯甲醇、n-甲基环己胺、混合催化剂和溶剂混合后进行缩合反应,生成溴己新;
(4)将所得溴己新和盐酸在溶剂中进行成盐反应,生成盐酸溴己新。
本发明将2-氨基苯甲酸酯类化合物、溴素、氧化剂和混合溶剂混合后进行溴代反应,生成3,5-二溴-2-氨基苯甲酸酯。
在本发明中,所述2-氨基苯甲酸酯类化合物优选包括2-氨基苯甲酸甲酯或2-氨基苯甲酸乙酯;当所述2-氨基苯甲酸酯类化合物为2-氨基苯甲酸甲酯时,溴代反应生成的为3,5-二溴-2-氨基苯甲酸甲酯;当所述2-氨基苯甲酸酯类化合物为2-氨基苯甲酸乙酯时,溴代反应生成的为3,5-二溴-2-氨基苯甲酸乙酯。
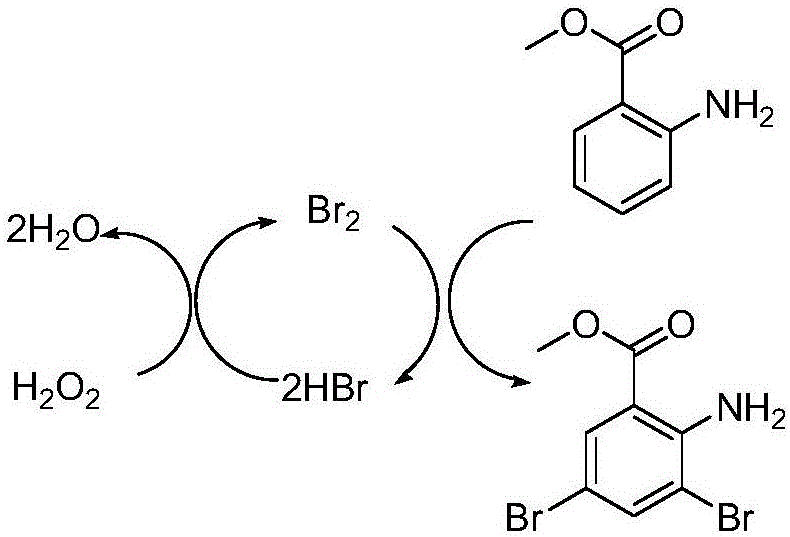
以所述2-氨基苯甲酸酯类化合物为2-氨基苯甲酸甲酯为例,所述溴代反应如式1所示,其反应原理如图1所示:
由图1可知,本发明实现了溴原子的100%利用,副产物为水,从而避免了溴化步骤中溴化氢的产生。此外,市场上的3,5-二溴-2-氨基苯甲酸酯大多含有氯代杂质,且该杂质很难除去。本发明提供的方法以液溴做溴源,能够避免接触氯原子而产生氯代杂质,无需酸性条件,不产生废酸,对环境十分友好,且得到的3,5-二溴-2-氨基苯甲酸酯不含氯代杂质,提高了产品的纯度和收率。
在本发明中,所述氧化剂优选包括过氧化氢、间氯过氧苯甲酸和过氧乙酸中的一种或几种,更优选为过氧化氢。在本发明具体实施例中,所述过氧化氢优选为30%的过氧化氢溶液。
在本发明中,所述混合溶剂优选为水和有机溶剂的混合物;所述有机溶剂优选为四氢呋喃、乙酸乙酯和二氯甲烷中的一种或几种,更优选为二氯甲烷;所述混合溶剂中水和有机溶剂的体积比优选为2:(2~6),更优选为2:(4~5)。
在本发明中,所述2-氨基苯甲酸酯类化合物和溴素的摩尔比优选为1:(1~2),更优选为1:(1.05~1.5);所述2-氨基苯甲酸酯类化合物和氧化剂的摩尔比优选为1:(1~2),更优选为1:(1.2~1.5);所述2-氨基苯甲酸酯类化合物和混合溶剂的质量比优选为1:(10~15),更优选为1:(12~13.5)。
本发明优选先将2-氨基苯甲酸酯类化合物溶于部分有机溶剂和全部水中,得到2-氨基苯甲酸酯类化合物溶液;将溴素溶于剩余的有机溶剂中,得到溴溶液;最后再将溴溶液加入到2-氨基苯甲酸酯类化合物溶液中,搅拌均匀得到反应物混合液。本发明优选在得到所述反应物混合液后再向其中加入所述氧化剂进行反应。本发明对溶解2-氨基苯甲酸酯类化合物和溴素的两部分有机溶剂的量的分配没有特殊要求,能够分别将两种物质溶解即可。在本发明中,所述溴代反应优选在室温下进行,所述溴代反应的时间优选为6~8h,更优选为7h。
所述溴代反应时间达到后,本发明优选向反应体系中缓慢加入亚硫酸钠溶液将反应淬灭;所述亚硫酸钠溶液的浓度优选为15~25%,更优选为20%。
淬灭反应后,本发明优选对得到的体系进行分相,分相得到的有机相用水萃取,分相得到的水相用有机溶剂(与溴代反应所选有机溶剂相同)萃取,合并有机相后将有机相减压浓缩至无大量溶剂馏出,将剩余物溶于甲醇中之后静置冷至室温,然后再冷却至0℃析晶2h。所述析晶结束后过滤得到固体,干燥后即得到纯净的3,5-二溴-2-氨基苯甲酸酯。
得到3,5-二溴-2-氨基苯甲酸酯后,本发明将所得3,5-二溴-2-氨基苯甲酸酯、催化剂、还原剂和溶剂混合后进行还原反应,生成3,5-二溴-2-氨基苯甲醇。
当所述3,5-二溴-2-氨基苯甲酸酯为3,5-二溴-2-氨基苯甲酸甲酯时,所述还原反应如式2所示:
在本发明中,所述催化剂优选为氯化钙、氯化锂和氯化锌中的一种或几种,更优选为氯化钙;所述还原剂优选为硼氢化钠、硼氢化锂或硼氢化钾中的一种或几种,更优选为硼氢化钾;所述溶剂优选为甲醇、无水乙醇和四氢呋喃中的一种或几种,更优选为无水乙醇。
在本发明中,所述3,5-二溴-2-氨基苯甲酸酯、催化剂和还原剂的摩尔比优选为1:(1.0~1.5):(2.0~3.0),更优选为1:(1.2~1.5):(2.0~2.5);所述3,5-二溴-2-氨基苯甲酸酯和溶剂的质量比优选为1:(3.5~4.5),更优选为1:4。
本发明优选先将3,5-二溴-2-氨基苯甲酸酯溶于部分溶剂中,然后再与还原剂混合得到反应液;将催化剂溶于剩余部分溶剂中得到催化剂溶液,然后在热环境下缓慢的将所得催化剂溶液加入到反应液中,加毕之后保温继续进行还原反应。在本发明中,所述热环境具体为将催化剂溶液加入到热的反应液中,所述反应液的温度为还原反应的温度;本发明加入所述催化剂溶液的时间优选≥6h。本发明对溶解3,5-二溴-2-氨基苯甲酸酯和催化剂的两部分溶剂的量的分配没有特殊要求,能够分别将两种物质溶解即可。在本发明中,所述还原反应的温度优选为35~45℃,更优选为40~42℃;时间优选为14~16h,更优选为14h。
本发明在所述还原反应过程中可取样进行uplc检测,当3,5-二溴-2-氨基苯甲酸酯含量不大于1.0%时视为反应合格。待反应合格后,本发明优选将反应体系冷却至室温,且维持反应温度不超过35℃,向反应体系中加入5%盐酸调节体系的ph值稳定在2~3。本发明优选对调节了ph值之后的体系进行离心过滤,用水将滤饼洗至中性。本发明优选将所得中性滤饼加入到0.4%氢氧化钠溶液中,在40~50℃下搅拌10~20min,更优选的在40℃下搅拌15min,之后冷至室温,离心过滤,再次水洗至中性,干燥,得到纯净的3,5-二溴-2-氨基苯甲醇。
得到3,5-二溴-2-氨基苯甲醇后,本发明将所得3,5-二溴-2-氨基苯甲醇、n-甲基环己胺、混合催化剂和溶剂混合后进行缩合反应,生成溴己新。
本发明所述缩合反应如式3所示:
在本发明中,所述混合催化剂优选包含超强酸和醋酸,所述超强酸优选为市售的sio2-al2o3、sio2-zro3、sio2-oso3h和sio2-tio2中的一种或几种,所述醋酸的质量浓度为36~38%,所述超强酸和醋酸的质量比优选为(0.08~0.15):(0.2~0.5),更优选为(0.1~0.12):(0.25~0.4)。在本发明中,所述3,5-二溴-2-氨基苯甲醇、n-甲基环己胺和混合催化剂的质量比优选为1:(0.9~1.0):(0.3~0.4),更优选为1:(0.95~0.98):(0.35~0.38)。
在本发明中,所述溶剂优选为甲苯、二甲苯和环己烷中的一种或几种,更优选为甲苯;所述3,5-二溴-2-氨基苯甲醇和溶剂的质量比优选为1:(0.01~0.2),更优选为1:(0.08~0.15)。
在本发明中,所述缩合反应的温度优选为80~90℃,更优选为85~88℃;时间优选为10~14h,更优选为12~13h。
在制备溴己新的缩合反应中,现有技术一般都需要在高温180℃下缩合大于20小时,该反应需要特殊设备,耗能大。而且高温导致api杂质增多。本发明采用的混合催化剂,超强酸催化剂与醋酸协同催化,可以降低反应温度,缩短反应时间。
得到溴己新后,本发明将所得溴己新和盐酸在溶剂中进行成盐反应,生成盐酸溴己新。
在本发明中,所述溶剂优选为甲醇、乙醇、异丙醇、乙酸乙酯和丙酮中的一种或几种,更优选为丙酮;所述溴己新和溶剂的质量比优选为1:(4~5),更优选为1:4.5。
在本发明中,所述盐酸的质量浓度优选为10~20%,更优选为15~18%;所述盐酸的添加量以使得体系的ph值达到2~3为准。
本发明优选将所述溴己新溶于溶剂中之后再添加盐酸析出盐酸溴己新固体;本发明可以通过搅拌方式来促进固体的析出。
在本发明中,所述步骤(4)中成盐反应的温度优选为0℃~室温,更优选为10~20℃;时间优选为4~6h。
所述成盐反应结束后,本发明优选对析出的盐酸溴己新粗品进行精制,所述精制优选包含如下步骤:
将所述盐酸溴己新粗品溶于溶剂中,得到盐酸溴己新溶液;
在回流条件下,向所述盐酸溴己新溶液中加入活性炭进行脱色;
将脱色得到的体系进行过滤,所述滤液顺次进行室温析晶和低温析晶,得到晶体产品;
将所述晶体产品顺次进行醇洗和干燥,得到纯净的盐酸溴己新。
在本发明中,所述溶剂优选为乙醇的水溶液,所述溶剂的质量浓度优选为95%;所述盐酸溴己新粗品和溶剂的质量比优选为1:12~15,更优选为1:14。
在本发明中,所述活性炭和盐酸溴己新粗品的质量比优选为0.01~0.1:1,更优选为0.05:1。
在本发明中,所述室温析晶优选在搅拌条件下进行,所述室温析晶的时间优选为1~3h,更优选为2h。
在本发明中,所述低温析晶优选在搅拌条件下进行,所述低温析晶的温度优选为3~7℃,更优选为5℃。
在本发明中,所述干燥优选为真空干燥,温度优选为55~65℃,更优选为60℃;时间优选为10~14h,更优选为12h;真空度优选小于等于-0.08mpa。
下面结合实施例对本发明提供的技术方案进行详细的说明,但是不能把它们理解为对本发明保护范围的限定。
实施例1
本实施例中的原料均来源于市售。
3,5-二溴-2-氨基苯甲酸甲酯的合成:
在100l双层玻璃反应釜中依次加入二氯甲烷26.50kg、纯化水8.0kg、2-氨基苯甲酸甲酯3.02kg,搅拌均匀。常温下,向上述所得反应液中缓慢加入溴素(3.83kg)的二氯甲烷(5.3kg)溶液。加入完毕后继续搅拌10分钟,向反应液中缓慢加入30%的过氧化氢溶液2.50kg,搅拌6小时反应完全后,向反应液中缓慢加入20%的亚硫酸钠溶液3.5kg淬灭反应,分相,有机相用8.0kg用纯化水萃取一次,水相用二氯甲烷萃取两次,合并有机相,将有机相减压浓缩至无大量溶剂馏出,将剩余物转至100l反应釜中并加入甲醇15.0kg,加热至溶解,搅拌15分钟。将反应液转至析晶桶中,静止缓慢冷却至室温,再放置冰箱中继续冷却至0℃析晶2小时。过滤得固体,50℃干燥,水分合格后收料5.88kg,收率为96%。
3,5-二溴-2-氨基苯甲醇的合成:
在100l双层玻璃反应釜中加入无水乙醇10.0kg、3,5-二溴-2-氨基苯甲酸甲酯5.88kg,搅拌均匀后再加入硼氢化钾2.62kg,室温下继续搅拌30分钟。将反应液升温至40℃,向反应液中缓慢加入含氯化钙2.7kg的无水乙醇溶液13.5g,加入时间不小于6小时。加毕,保温搅拌反应8小时,取样uplc检测。反应合格后(3,5-二溴-2-氨基苯甲酸甲酯均不大于1.0%),冷却至室温,维持反应液温度不超过35℃,向反应液中缓慢加入32kg5%的盐酸调节ph至2~3,加完继续搅拌20分钟,且复测ph不变。离心过滤,用纯化水淋洗滤饼至中性,将所得固体转至反应釜中,加入0.4%的氢氧化钠溶液20kg,加热至45℃搅拌15分钟,然后冷却至室温。离心过滤,用纯化水淋洗滤饼至中性。平铺于鼓风干燥箱托盘中,60℃,干燥20小时,水分合格后收料5.08kg,收率95%。
盐酸溴己新粗品制备:
30l带分水器的玻璃反应釜中加入n-甲基环己胺4.84kg、甲苯0.44kg、3,5-二溴-2-氨基苯甲醇5.0kg,搅拌下加入固体超强酸sio2-oso3h(ssa)0.5kg,醋酸1.2kg,然后升温至85℃,总共反应12小时后,取样uplc检测(取反应液为样品,浓度约为275mg/ml)。反应合格后(3,5-二溴-2-氨基苯甲醇不大于5.0%),减压浓缩至无明显馏份馏出,剩余物转至桶中冷却至室温后加入丙酮12kg。然后将溶液转至100l双层玻璃反应釜中,用15%的盐酸调节至ph2~3析出固体,20分钟后,复测ph2~3;继续搅拌1小时,然后继续冷却至0℃搅拌4小时,离心过滤,得盐酸溴己新湿品,将湿品平铺于鼓风干燥箱托盘内,35±5℃干燥,每3小时翻料1次,干燥10小时,水分合格后收料6.24kg,收率85%。
盐酸溴己新精制:
在100l搪瓷反应釜中加入95%乙醇42.0kg、盐酸溴己新粗品6.2kg、升温至回流,搅拌溶解后再加入活性炭0.15kg,搅拌脱色1小时。钛棒过滤,滤液转至100l双层玻璃反应釜中,缓慢降温至室温,然后室温搅拌析晶2小时,再降温至5℃搅拌析晶5小时。离心过滤,滤饼用少量95%乙醇淋洗,得到盐酸溴己新湿品。将盐酸溴己新湿品平铺于真空干燥箱托盘内,60℃干燥,真空度小于-0.08mpa,每2小时翻料一次,干燥12小时,水分合格后收料4.96kg,收率80%。
本发明所得盐酸溴己新的结构如式4所示:
按照《中国药典》(2015年版,二部)盐酸溴己新【检查】项下有关物质的测定方法对所得盐酸溴己新进行杂质测定,测定杂质的含量,其uplc谱图如图2所示。结果显示未检测到杂质,纯度100%。
图2相关的样品信息如表1.1和表1.2所示。
表1.1
表1.2
将本发明所得盐酸溴己新和氯代杂质(杂质i:
图3相关的样品信息如表2.1和表2.2所示。
表2.1
表2.2
本发明所得盐酸溴己新的结构检测数据和谱图分别如表3~5和图4~5所示,其中,表3为盐酸溴己新的1hnmr数据,图4为1hnmr谱图;表4为盐酸溴己新的13cnmr数据,图5为13cnmr谱图;表5为盐酸溴己新的xrd数据。由表3~5和图4~5可知,本发明提供的盐酸溴己新为目标产物。
表3盐酸溴己新的1hnmr数据
表4盐酸溴己新的13cnmr数据
表5盐酸溴己新的xrd数据
实施例2
将实施例1中的3.02kg2-氨基苯甲酸甲酯替换为3.3kg2-氨基苯甲酸乙酯,其它条件完全一致,最终能够成功制得盐酸溴己新,且各步骤收率相当。
以上所述仅是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。
- 还没有人留言评论。精彩留言会获得点赞!