一种测序文库构建试剂盒及其使用方法和应用与流程
本发明涉及基因检测领域,具体涉及一种测序文库构建试剂盒及其使用方法和应用。
背景技术:
拷贝数变异(copynumbervariation,cnv)是指基因组片段发生了拷贝数的增加或减少,涉及的基因组序列大小从1kb到多个mb不等,主要是由于基因组发生重组而导致。cnv是一种广泛存在于动物基因组中的遗传多态性现象,它的突变频率远高于snp。有些cnv不引起表型变化,而另一些cnv则会影响基因的表达与功能,最终导致人类疾病的发生,因此cnv的研究具有重大的科研和临床意义。
目前检测cnv的技术主要有:高分辨率核型分析技术,免疫荧光原位杂交技术(fluorescentinsituhybridization,fish),染色体微阵列分析技术(chromosomemicroarrayanalysis,cma),多重连接探针扩增技术(multiplexligation-dependentprobeamplification,mlpa)以及二代测序技术(next-generationsequencing,ngs)。
高分辨率核型分析技术是检测染色体异常的金标准,但是其分辨率较低(约5mb),无法检出染色体小片段的拷贝数变异。免疫荧光原位杂交技术利用荧光标记探针和染色体进行原位杂交,根据特定的同位素荧光信号在分子水平上检测染色体数目和结构异常。相对于核型分析技术,fish检测周期更短,精度更高,但是由于探针的设计受到目标区域的限制,只能得到有限的染色体信息。染色体微阵列分析技术(cma)是一种高通量检测基因组拷贝数变异的分子核型技术,包括比较基因组杂交(acgh)技术和单核苷酸多态性基因芯片(snparray)技术。相比于fish,cma具有更高的分辨率和灵敏度,全基因组覆盖,不过均一性不好,容易漏检,且检测成本较高,限制了在临床的应用。mlpa是一项基于pcr的检测cnv变化的技术,可以检测出dna特异性序列间的拷贝数差别。
mlpa可以对一个样本的几十上百个位点进行同时检测,不过操作过程繁琐,位点依然有限,容易造成pcr产物的污染。
技术实现要素:
针对现有技术中存在的不足,本发明所提供的技术方案是:
一种测序文库构建试剂盒,具体包括:限制性内切酶及限制性内切酶反应的缓冲液,dna连接酶及连接酶缓冲液,末端修复试剂,测序接头,pcr引物以及pcr扩增体系;
所述末端修复试剂包括t4多聚核苷酸激酶,t4dna聚合酶,klenowdna聚合酶,dntps和修复缓冲液。
所述限制性内切酶试剂包括asei,cviqi,ndei,aciⅰ,aluⅰ,bfaⅰ,haeⅲ,hhaⅰ,mseⅰ,asisi,cviaii,paci,pvui,pvui-hf,hpych4iv或hpyse526i中的一种或几种。
所述dna连接酶试剂包括dnaligase、t4dnaligase、taqdnaligase或ligase-65的一种。
所述pcr引物包括上游引物和下游引物。
所述pcr扩增体系包括dna聚合酶,dntps,扩增缓冲液。
本发明还包括一种测序文库构建试剂盒的使用方法,具体包括以下步骤:
将样本基因组dna进行片段化处理,获得第一dna片段组;
在所述第一dna片段组加入末端修复试剂,置于pcr仪上进行反应获得末端修复的第二dna片段组;
将所述第二dna片段组与dna连接酶,测序接头,连接酶缓冲液,无核酸酶去离子水混合,置于pcr仪上反应获得接头连接dna片段组,并对所述接头连接dna片段组进行纯化;
将所述纯化后的接头连接dna片段组加入pcr引物和pcr扩增体系,在预定的扩增程序下进行扩增获得扩增基因组,并对所述扩增基因组进行纯化;
将所述纯化后的扩增基因组进行检测,获得测序文库。
所述将样本基因组dna进行片段化处理,获得第一dna片段组的方法,具体包括以下步骤:
将样品基因组dna与限制性内切酶,限制性内切酶缓冲液混合置于pcr仪上反应获得第一酶切dna片段组;
将所述第一酶切dna片段组与dna连接酶,连接酶缓冲液,无核酸酶去离子水混合置于pcr仪上反应获得第一连接dna片段组,并采用磁珠纯化;
将所述纯化后的第一连接dna片段组采用物理法或者酶解法打断获得第一dna片段组。
本发明还提供了另一种将样本基因组dna进行片段化处理,获得第一dna片段组的方法,具体包括以下步骤:
将样品基因组dna采用物理法打断,并加入限制性内切酶和限制性
内切酶缓冲液混合均与,置于pcr仪上反应获得第二酶切dna片段组;
将第二酶切dna片段组与dna连接酶,连接酶缓冲液,无核酸酶去离子水混合置于pcr仪上反应获得第二连接dna片段组,并进行纯化;
所述纯化后的第二连接dna片段组为第一连接dna片段组。
所述预定的扩增程序包括以下步骤:
95℃预变性3分钟;
循环反应,95℃变性20秒,60℃退火15秒,72℃延伸30秒,共循环4-8次;
72℃延伸5分钟。
有益效果:
利用本发明的测序文库构建试剂盒,将基因组dna进行片段化出里,末端修复,接头连接、纯化等步骤构建测序文库,在进行拷贝数变异测序时,双端测序的read1和read2的序列很大程度上来自于基因组上的不同位置(间隔>500bp以上),使得用来分析基因组拷贝数变异时,read1和read2都能得以利用,提高原有数据的利用效率150%以上。
附图说明
图1为本发明实施例二提供的一种测序文库构建方法的原理图;
图2为本发明实施例二第一酶切dna片段组和第一连接dna片段组的琼脂糖凝胶电泳图;
图3为本发明实施例三提供的一种测序文库构建方法的原理图;
图4为本发明实施例三中的第二酶切dna片段组的琼脂糖凝胶电泳图;
图5为本发明实施例三中的第二连接dna片段组的琼脂糖凝胶电泳图;
具体实施方式
为了更加清楚阐述本发明的技术内容,在此结合具体实施例予以详细说明,显然,所列举的实施例只是本技术方案的优选实施方案,本领域的技术人员可以根据所公开的技术内容显而易见地得出的其他技术方案仍属于本发明的保护范围。
原理解释:
利用二代高通量测序数据进行cnv分析的一个主要思路是根据一定大小的滑动窗口(window,比如1mb)内读段深度(用cluster提供的坐标信息的个数来表示)来指示拷贝数增加(duplication)与缺失(deletion),这种分析原则得以成立的前提是测序文库中的dna片段在基因组上的分布符合泊松分布现象,二代高通量测序中每条dna序列信号来源的起点叫做cluster,每个cluster的信息通常由read1和read2组成,read1和read2通过将呈现出来的序列信息与参考基因组进行比对,就能获得read1和read2在参考基因组上的坐标信息。
实施例1:
本发明提供了一种用于测序文库构建的试剂盒,具体包括限制性内切酶,限制性内切酶缓冲液,末端修复试剂,dna连接酶试剂组,连接酶缓冲液,测序接头(01-96),pcr引物组,pcr反应体系。
其中限制性内切酶为asei,cviqi,ndei,aciⅰ,aluⅰ,bfaⅰ,haeⅲ,hhaⅰ,mseⅰ,asisi,cviaii,paci,pvui,pvui-hf,hpych4iv,hpyse526i中的一种或几种的组合,应当理解的是可以根据测序文库针对的对象选择不同的限制性内切酶,可以包括但不限于上述所列出的限制性内切酶。
其中末端修复试剂组包括t4pnk(t4polynucleotidekinase,t4多聚核苷酸激酶),t4dnapolymerase(t4dna聚合酶),klenowdnapolymerase(klenowdna聚合酶),dntps,修复缓冲液。
其中dna连接酶试剂组中连接酶可以是dnaligase、t4dnaligase、taqdnaligase或ligase-65。
其中测序接头为人工合成的寡核苷酸dna片段,共包括测序接头01-96共96种,购自南京诺唯赞生物科技有限公司。
pcr引物(南京诺唯赞生物科技有限公司)包括上游引物p5,是文库p5端接头特异性引物序列,下游引物p7,是文库p7端接头特异性引物序列。p5核酸序列为:5'-aatgatacggcgaccaccgaga-3';p7核酸序列为:p7:5'-caagcagaagacggcatacga-3'。
其中pcr反应体系为hifi扩增混合物,包括dna聚合酶,dntps,扩增缓冲液。
实施例2
附图1示出了一种测序文库构建方法的原理图,详述如下:
原理步骤1-1:样本基因组不同染色体被限制性内切酶试剂组切割后,形成大小不一的片段,这些片段都具有可供再次连接的5'-磷酸基团和3'-羟基基团;
原理步骤1-2:在dna连接酶试剂组存在的情况下,不同的dna片段随机连接,再次成为长片段;
原理步骤1-3:利用超声打断等随机打断方式,将重新形成的长片段再次随机打断,得到含有来自不同区域的片段;
原理步骤1-4:与测序接头进行连接后,形成完整的可供扩增的文库dna片段;
原理步骤1-5:在测序时,同一个测序簇cluster中,至少会有两种不同的情况,比如测序簇cluster1中,read1的信息来自chr2-3,部分来自chr17-4,read2的信息来自chr6-2,部分来自chr17-4,在cnv分析中,该read1和read2的信息一共能提供三个有效物理位置信息,而在测序簇cluster2中,read1和read2都来自chr17-2,实际上只为cnv提供了一个有效物理位置信息;传统的建库方式的每个cluster只为cnv分析提供一个有效物理位置信息,而本专利提供的建库方式的同一个cluster有能力为cnv分析提供更多有效物理位置信息,因此,本发明所提供的建库方式提高了测序信息的利用效率,使得能在相同的测序数据量情况下,为cnv分析提供更多有效物理位置信息。
具体实施方式如下:
首先提取基因组dna本血液抽提试剂盒购自天根生化科技有限公司,提取方法包括:
取200μl血液样本到2.0ml的离心管中。若提取小于200μl血液样品时,可加入缓冲液gs补足体积至200μl,再进行下一步实验。
加入200μlbuffergb和20μl蛋白酶k溶液至上述样品中,充分振荡混匀。
在56℃孵育10min,期间颠倒混匀数次,溶液应变清亮(若溶液未彻底变清亮,请延长裂解时间至溶液清亮为止)。
室温放置2-5min后加入350μlbufferbd,充分颠倒混匀,短暂离心将反应液收集至管底。
将所有溶液分转移到离心柱中(离心柱放入收集管中),12000rpm(~13400×g)离心30sec,弃废液,将离心柱放回收集管中。
加入500μlbuffergdb,12000rpm(~13400×g)离心30sec,弃废液,将离心柱放回收集管中。
加入600μlbufferpwb,12000rpm(~13400×g)离心30sec,弃废液,将离心柱放回收集管中。
重复上一步骤。
12000rpm(~13400×g)离心2min,将离心柱转入一个新的1.5ml离心管中,开盖室温放置5min,以彻底风干吸附膜上的残余的缓冲液。
向离心柱的吸附膜中心悬空加入50μl洗脱液te,室温放置2min,12000rpm(~13400×g)离心2min,将洗脱液收集到离心管中。
用nanodrop测定dna浓度和a260/a280的比值。
在抽提合格的dna34μl(共500ng)溶液中加入4μl剪切酶缓冲液,1μlmsei(neb),1μlbfai(neb)置于pcr管中混合均匀,置于pcr仪上37℃反应1小时,之后65℃反应10分钟,获得第一酶切dna片段组。
取5μl上述产物进行电泳检测,电泳图详见图2.
在第一酶切dna片段组中加入5μl连接酶缓冲液,2μldna连接酶以及8μl无核酸酶去离子水,混合均匀后,置于pcr仪上20℃反应2小时获得第一连接dna片段组。
取5μl上述产物进行电泳检测。
用1倍体积ampurexpbeads(核酸纯化试剂盒)对余下第一连接dna片段组进行纯化并用51μl无核酸酶去离子水进行洗脱。
电泳图如图2所示。其中泳道1泳道为1500bpmarker,片段长度由上而下依次为1500bp,1000bp,900bp,800bp,700bp,600bp,500bp,400bp,300bp,200bp,100bp;2-4泳道为基因组dna经限制性内切酶酶切后的产物,泳道5-6为限制性内切酶酶切的产物经过dna连接酶连接后的产物。
利用covariss220对纯化后的第一连接dna片段组进行片段化处理,将covariss220仪器的水槽内注入去离子水,至水位达到12,依次打开covariss220仪器,变压器以及制冷循环系统的开关,打开软件,当前视图显示制冷循环为off,点击enter,此时屏幕显示当前水温,制冷循环系统开始工作,水槽开始排气,取第一连接dna片段组缓慢加入至干净的covarismicrotube中,短暂离心,小心地将covarismicrotube放入s220的支撑架,确保tube保存竖直方向。根据表1的条件设置对dna进行超声打断。
表1
从covarismicrotube中将片段化后获得第一dna片段组转移到一个新的pcr管中。
往第一dna片段组中加入15μl末端修复试剂组,混合均匀后,置于pcr仪上20℃反应15分钟,之后65℃反应15分钟。
往末端修复产物中加入10μl连接酶缓冲液,4μldna连接酶,测序接头5μl和16μl无核酸酶去离子水,混合均匀后,置于pcr仪上20℃反应15分钟获得接头连接dna片段组。
用0.6倍体积ampurexpbeads对接头连接dna片段组进行纯化,并用21μl无核酸酶去离子水进行洗脱。
取纯化产物20μl于pcr管中,加入pcr引物组5μl和hifi扩增体系混合物25μl,混合均匀后,置于pcr仪进行下述反应:95℃预变性3分钟,接着进入循环反应,95℃变性20秒,60℃退火15秒,72℃延伸30秒,共4个循环后,继续72℃延伸5分钟。
用0.9倍体积ampurexpbeads对扩增产物进行纯化,并用21μl无核酸酶去离子水进行洗脱。然后用基于双链dna荧光染料的方法或基于qpcr绝对定量的方法对文库浓度进行检测,所得文库即为最终上机文库。
数据分析:
测试利用上述试剂盒构建的测序文库中的四个样本,测序获得3.36-4.08mclusters,并对其进行每个cluster含有参考基因组不同坐标位置的可能性和比例的分析,例如lc19样本,测序总共获得4,078,359个clusters,其中能产生1、2、3、4和>4个有效物理位置信息的clusters占比分别是32.287%,43.265%,17.715%,4.448%,0.804%,总体数据有效提升159.825%,相当于原来只能利用4,078,359个有效物理位置信息,经过本实施例的建库方案和分析之后,有效物理位置信息总数增加到6,518,221个。具体数据如表2。
表2
由表2可知,利用本实施例的测序文库构建方法构建测试文库,用于cnv分析时,能够产生多个有效物理位置,使得有效物理位置总数大大增加,有效数据提升可达159%以上,使得测序效率大大增加。
实施例3
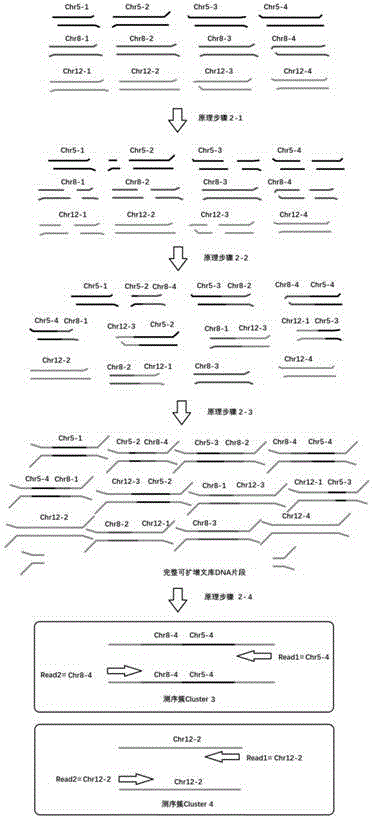
附图3示出了一种测序文库构建方法的原理图,详述如下:
原理步骤2-1:样本基因组dna先被超声打断,形成末端需要修复、不易连接的大小不一的片段;
原理步骤2-2:加入限制性酶切试剂组,含有内切酶识别位点的dna片段会被切断,形成具有5'-磷酸基团和3'-羟基基团的可以被连接的端口;
原理步骤2-3:在dna连接酶试剂组存在的情况下,不同的具有可以被连接端口的dna片段随机连接,形成一个新的杂合片段;不含内切酶识别位点、没有被限制性内切酶切断的片段,不会发生连接;
原理步骤1-4:经过末端修复和与测序接头进行连接后,形成完整的可供扩增的文库dna片段;
原理步骤1-5:在测序时,同一个测序簇cluster中,至少会有两种不同的情况,比如测序簇cluster3中,read1的信息来自chr5-4,read2的信息来自chr8-4,在cnv分析中,该read1和read2的信息各自提供了一个有效物理位置信息,而在测序簇cluster2中,read1和read2都来自chr12-2,实际上只为cnv分析提供了一个有效物理位置信息;传统的建库方式的每个测序簇只能为cnv分析提供一个有效物理位置信息,而本实例提供的建库方式的同一个cluster有能力为cnv分析提供更多的有效物理位置信息,因此,本实例所提供的建库方式提高了测序信息的利用效率,使得能在相同的测序数据量情况下,为cnv分析提供更多的有效物理位置信息。
具体实施方式如下:
按照实施例2的提取方法获得基因组dna。
将covariss220仪器的水槽内注入去离子水,至水位达到12,依次打开covariss220仪器,变压器以及制冷循环系统的开关,打开软件,当前视图显示制冷循环为off,点击enter,此时屏幕显示当前水温,制冷循环系统开始工作,水槽开始排气。
取基因组dna缓慢加入至干净的covarismicrotube中,短暂离心,小心地将covarismicrotube放入s220的支撑架,确保tube保存竖直方向根据表3的条件设置对dna进行超声打断。
表3
从covarismicrotube中将片段化后的基因组dna转移到一个新的pcr管中。
取上述采用超声片段化后的基因组dna34μl,加入4μl剪切酶缓冲液,1μlmsei(neb)和1μlhhai(neb)于pcr管中混合均匀,置于pcr仪上37℃反应1小时,之后65℃反应10分钟获得第二酶切dna片段组,取5μl第二酶切dna片段组进行电泳检测。详见图4,其中泳道4泳道为1500bpmarker,片段长度由上而下依次为1500bp,1000bp,900bp,800bp,700bp,600bp,500bp,400bp,300bp,200bp,100bp;1-3泳道为片段化的基因组dna经限制性内切酶酶切后的产物。
在剩余的第二酶切dna片段组加入5μl连接酶缓冲液,2μldna连接酶以及8μl无核酸酶去离子水,混合均匀后,置于pcr仪上20℃反应2小时获得第二连接dna片段组。取5μl上述第二连接dna片段组进行电泳检测。详见图5,其中泳道1泳道为1500bpmarker,片段长度由上而下依次为1500bp,1000bp,900bp,800bp,700bp,600bp,500bp,400bp,300bp,200bp,100bp;2-4泳道为限制性内切酶酶切的产物经过dna连接酶连接后的产物。
用1倍体积ampurexpbeads对第二连接dna片段组进行纯化并用16μl无核酸酶去离子水进行洗脱。
向纯化后的第二连接dna片段组中加入15μl末端修复试剂,混合均匀后,置于pcr仪上20℃反应15分钟,之后65℃反应15分钟获得末端修复的第二dna片段组。
往末端修复的第二dna片段组中加入10μlligationbuffer,4μldnaligase,测序接头5μl和16μlnucleasefreewater,混合均匀后,置于pcr仪上20℃反应15分钟获得接头连接dna片段组。用0.6倍体积ampurexpbeads对接头连接dna片段组进行纯化,并用21μl无核酸酶去离子水进行洗脱。
取纯化后的接头连接的dna片段组20μl于pcr管中,加入pcr引物组5μl和hifi反应体系混合物25μl,混合均匀后,置于pcr仪进行下述反应:95℃预变性3分钟,接着进入循环反应,95℃变性20秒,60℃退火15秒,72℃延伸30秒,共4个循环后,继续72℃延伸5分钟。
用0.9倍体积ampurexpbeads对扩增产物进行纯化,并用21μl无核酸酶去离子水进行洗脱。然后用基于双链dna荧光染料的方法或基于qpcr绝对定量的方法对文库浓度进行检测,所得文库即为最终上机文库。
数据分析
测试利用上述试剂盒构建的测序文库中的四个样本,获得了3.06~9.12mclusters数据,并对其进行每个cluster含有参考基因组不同坐标位置的可能性和比例的分析,例如a2435样本,测序总共获得5,178,126个clusters,其中能产生1、2、3、4和>4个有效物理位置信息的clusters占比分别是31.692%,40.334%,20.555%,5.635%,0.907%,总体有效数据提升163.486%,相当于原来只能利用5,178,126个有效物理位置信息,经过本实施例的建库方案和分析之后,有效物理位置信息总数增加到8,465,497个。详见表4。
由表4可知,利用本实施例的测序文库构建方法构建测试文库,用于cnv分析时,能够产生多个有效物理位置,使得有效物理位置总数大大增加,有效数据提升可达161%以上,使得测序效率大大增加。
表4
随着测序数据量的增加,只能给一个有效物理位置信息的cluster的比例下降,比如3-5mclusters的样本(包括实施例2中的4个样本),只能提供一个有效物理位置信息的clusters比例在31~33%之间,而8~9mclusters的样本(本实施例汇总的a2436和a2437),只能提供一个有效物理位置信息的clusters比例减低到25~27%之间,其他类型的clusters比例都有所提升,总体有效数据提升大于170%。
根据上述说明书的揭示和教导,本发明所属领域的技术人员还可以对上述实施方式进行适当的变更和修改。因此,本发明并不局限于上面揭示和描述的具体实施方式,对本发明的一些修改和变更也应当落入本发明的权利要求的保护范围内。此外,尽管本说明书中使用了一些特定的术语,但这些术语只是为了方便说明,并不对本发明构成任何限制。
- 还没有人留言评论。精彩留言会获得点赞!