一种维生素K1的纯化工艺的制作方法
技术领域:
:本发明涉及一种原料药的纯化方法,特别涉及一种维生素k1的纯化工艺。
背景技术:
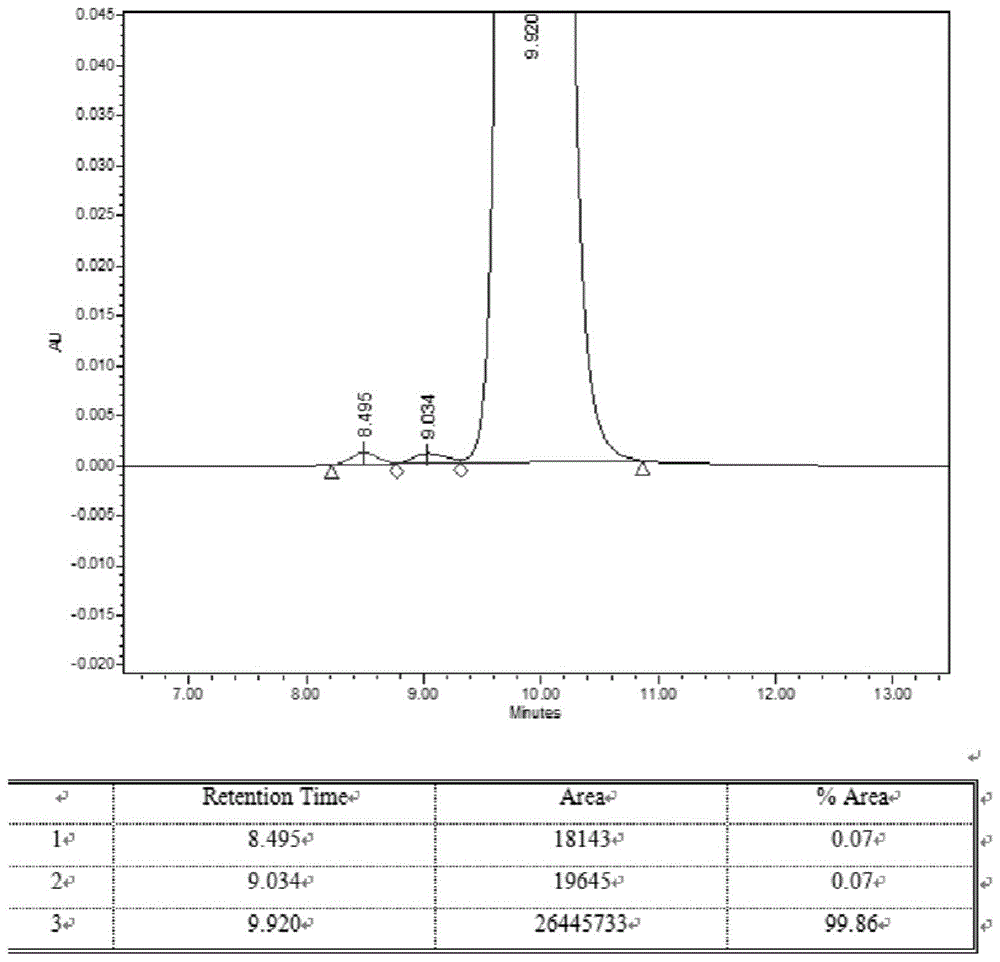
::维生素k1,英文名vitamink1,别名:植物甲萘醌;2-甲基-3-(3,7,11,15-四甲基十六-2-烯基)-1,4-萘醌,分子式c31h46o2,分子量450.71,结构式:维生素k1为维生素类及矿物质缺乏症用药。维生素k1注射液是2009版国家基本药物目录品种,主要用于各种维生素k缺乏引起的出血性疾病的治疗。在临床上应用于凝血酶过低症、维生素k1缺乏症、新生儿自然出血症的防治以及梗阻性黄疸、胆瘘、慢性腹泻等所致出血,香豆素类、水杨酸钠等所致的低凝血酶原血症。维生素还具有镇痛、缓解支气管痉挛的作用,对内脏平滑肌绞痛、胆管痉挛、肠痉挛引起的绞痛有明显的效果。维生素k1还可以用于多维食品和禽畜饲料的添加剂。维生素k1原料药为黄色至橙黄色透明粘稠的液体。无臭或几乎无臭;遇光易分解。在氯仿、乙醚或植物油中易溶,在乙醇中略溶,在水中不溶,折光率为1.525~1.528。2011年,国家食品药品监督管理局发布了第43期药品不良反应信息通报,表示维生素k1注射液存在严重过敏反应,提醒医务人员和患者要关注过敏反应的风险。数据分析显示,维生素k1注射液的安全风险除其本身外,还包括原料药中的杂质成分。维生素k1的制备方法包括:使用氧化银将未酯化的二氢维生素k1转化为维生素k1。使用六硝酸联铵为催化剂对二氢维生素k1二甲醚的氧化去甲基化可制备维生素k1。通过氧化二氢维生素k1碱金属盐制备维生素k1的方法。使用碱金属氢氧化物皂化二氢维生素k1单乙酸酯或单苯甲酸酯,然后使用空气氧化。日本专利公开(公开)212142/1982公开了使用二乙酯代替单酯为原料。使用空气氧化制备维生素k1方法。日本专利公开(公开)76854/1973公开了使用氧化银、氧化铅、二氧化锰、过氧化镍和二甲亚砜将二氢维生素k1氧化为维生素k1的方法现有方法所得到的产物,可能产生的杂质包括:维生素k1顺式异构体杂质(杂质3),溶剂残留,以及相关的过氧化产物或中间体,如"β,γ-dihydrovitamink1"(杂质2),"vitamink12,3-epoxide"(杂质1)等。对维生素k1的纯化问题是制备维生素k1原料药的难题,特别是如何去除上述杂质,现有技术报道极少。本发明人经过研究发现采用层析法有利于安全性的提高,但如何将其应用于工业化生产,仍需要探索。层析法是利用不同物质理化性质的差异而建立起来的技术。层析系统由两个相组成:一是固定相,另一是流动相。当待分离的混合物随流动相通过固定相时,由于各组分的理化性质存在差异,与两相发生相互作用(吸附、溶解、结合等)的能力不同,在两相中的分配(含量比)不同,且随流动相向前移动,各组分不断地在两相中进行再分配。分部收集流出液,可得到样品中所含的各单一组分,从而达到将各组分分离的目的。常用的层析方法包括,凝胶层析,离子交换层析,高效液相层析(hplc),亲和层析等。《沈阳药科大学学报》2013年第04期“维生素k1微乳剂在大鼠体内的药物动力学”孙婷;唐丽英;徐静瑶;赵云丽;于治国等公开了大鼠血浆中维生素k1的高效液相色谱测定法,并应用于经灌胃给予维生素k1微乳剂后的维生素k1药动学研究。方法为采用无水乙醇一乙醚(体积比为1:3)提取的方法处理血浆样品。采用反相高效液相色谱法测定维生素k1的血药浓度,色谱柱为thermoc18柱(150mm×4.6mm,5p.m),流动相为无水乙醇一水(体积比为90:10),流速为1.1ml·min-1,检测波长为270nm,柱温为35℃。2005版中国药典公开了维生素k1的检测方法:照高效液相色谱法(附录ⅴd)测定。避光操作。色谱条件与系统适用性试验用硅胶为填充剂;以石油醚(60-90℃)-正戊醇(2000:2.5)为流动相;检测波长为254nm。测定法取本品约20mg,精密称定.置50ml量瓶中.用流动相溶解并稀释至刻度,摇匀,精密量取该溶液5ml与内标溶液1ml,置10ml量瓶中,以流动相稀释至刻度.摇匀.取10μl注入液相色谱仪,记录色谱图;另取维生素k1对照品适量,精密称定,同法测定。按内标法以顺、反式异构体峰面积的和计算,即得。本发明人对现有技术进行了研究后,找到一种适合维生素k1的层析方法。技术实现要素::本发明提供一种维生素k1原料药的纯化方法,所述方法,包括以下步骤:步骤1,使用c18反相硅胶柱作为填料的dac600制备色谱柱,甲醇和乙酸乙酯混合溶液为流动相,对合成得到的vk1粗品进行层析纯化。步骤2,步骤1收获的经过反相硅胶柱层析纯化的vk1进一步用正相层析硅胶作为填料,正己烷为流动相,对vk1进行层析纯化。其中,步骤1的层析条件如下:流动相为:甲醇和乙酸乙酯,两者比例为200:85,检测波长:270nm,300nm,流速:10l/min,上样量:5%,样品浓度:400mg/ml步骤1的操作过程如下:1)流动相配制将甲醇1781kg和乙酸乙酯676kg加入反应釜并混合搅拌均匀。2)样品配制称取vk1粗品9.00kg,加入乙酸乙酯8.00kg搅拌溶清,将vk1粗品的乙酸乙酯溶液过预柱过滤以除去机械杂质和活性炭,用剩余的2.22kg乙酸乙酯冲洗,冲洗液合并到滤液中,搅拌下称量加入甲醇8.88kg,搅拌并保证溶液为澄清状态;一般甲醇用量会比乙酸乙酯少(7.20kg左右),否则会发生浑浊;若溶液发生浑浊,则向其中加入少量乙酸乙酯即可溶清(0.20kg左右);将配制好的样品溶液平均分成4份待用。3)进针,冲柱,进针,洗脱;4)洗脱液浓缩将所收集的洗脱液减压浓缩,蒸出溶剂,加热从30℃逐渐升温至70℃,减压真空≥0.095mpa,抽蒸5~10小时,检测折光率1.520~1.528之间,出料,得到vk1精制品,供正相层析精制。其中,步骤2的层析条件如下:溶剂和洗脱液:正己烷填料:正相层析硅胶+活性炭步骤2操作过程如下:1)备柱取硅胶,加入正己烷浸泡装入层析柱中;再在硅胶上层铺垫一层活性炭;2)装料vk1精制品加入正己烷溶解,打开层析柱底部料液出口,使料液进入柱中;3)洗脱加入正己烷洗脱液,进行洗脱。流速大约在2~3kg/min;前洗脱无色溶液返回流动相釜继续使用,待洗脱出黄色料液,开始收集,收集时间约2~3小时;4)浓缩将所收集的层析液减压浓缩,得vk1纯品。经过检测,本发明vk1纯品,各种指标如下:采用现有方法制备得到的vk1,有关杂质含量如下:收率总杂含量杂质1含量杂质2含量杂质3含量样品50%0.250.06%0.12%0.07%其中,现有方法为采用正相硅胶柱,流动相为3%乙酸乙酯的石油醚溶液,不加压自然过滤洗脱,重复2次过柱分离,单次过柱耗时约10天,单次溶剂消耗每公斤原料药约1200升。注:其中所述含量为重量百分含量本发明的优点:1、反相柱采用dac600型工业自动化制备柱,单次分离时间约30~40分钟;正相柱采用传统的正相硅胶柱,正己烷为流动相,加压快速过滤分离,单次分离时间约2~3小时,节省时间和人工成本;2、反相柱流动相消耗每公斤原料药约300升,正相柱流动相消耗每公斤原料药120升,较传统硅胶柱分离纯化大大节省了溶剂消耗。3、反相柱和正相柱全部采用目标物段全接收方法,单次过柱收率80%以上,总收率65%左右,操作简单,适合工业化生产。本发明的核心技术在于步骤1,其中的层析条件是经过筛选获得的,筛选过程如下:流动相的筛选1、反相柱流动相可用甲醇和乙酸乙酯、乙醇和乙酸乙酯、甲醇和二氯甲烷、乙醇和二氯甲烷等体系,其中乙醇和乙酸乙酯从分离效果和溶剂安全方面为最佳选择;2、正相柱流动相可用石油醚、正己烷、异辛烷等烷烃类溶剂,其中正己烷效果最佳。色谱柱的筛选1、反相柱为dac600工业制备柱,填料选用c18硅胶,c18硅胶对于化物的保留能力强于c8硅胶,适合vk1这种弱极性化合物的分离;2、正相柱采用100到200目硅胶做为填料。附图说明:图1本发明实施例1所得vk1高效液相色谱图,检测方法参照药典。其中1为杂质1,2为杂质2,3为vk1,被测样品为第一批样品1。具体实施方式:以下通过实施例进一步说明本发明,但不作为对本发明的限制。实施例1vk1柱层析工艺(单批粗品9kg批量)一、工序一:dac600柱层析(c18反相硅胶柱)1、物料和设备信息1-1物料配比物料名称配比(w/w)投料量(kg)vk1粗品19.00甲醇2002000乙酸乙酯94.448501-2设备信息2、实验条件检测波长:270nm,300nm流速:10l/min流动相:甲醇:乙酸乙酯上样量:5%样品浓度:400mg/ml3、操作过程1)流动相配制将甲醇1781kg和乙酸乙酯676kg加入反应釜并混合搅拌均匀。2)样品配制称取vk1粗品9.00kg,加入乙酸乙酯8.00kg搅拌溶清,将vk1粗品的乙酸乙酯溶液过预柱过滤以除去机械杂质和活性炭,用剩余的2.22kg乙酸乙酯冲洗,冲洗液合并到滤液中,搅拌下称量加入甲醇8.88kg,搅拌并保证溶液为澄清状态;一般甲醇用量会比乙酸乙酯少(7.20kg左右),否则会发生浑浊;若溶液发生浑浊,则向其中加入少量乙酸乙酯即可溶清(0.20kg左右);将配制好的样品溶液平均分成4份待用。3)平衡仪器用配制好的甲醇与乙酸乙酯混合溶液冲洗柱子至检测基线平衡,流速10l/min。3)进第一针(1)将一份样品溶液用流动相泵抽入色谱柱,流速5l/min;用甲醇和乙酸乙酯配制的流动相清洗样品瓶,清洗液一并抽入色谱柱以清洗进样管路,清洗标准为泵头前段透明管路中无明显颜色即可;(2)样品进入柱后,按照设定好的方法进行洗脱,当vk1峰升起即可开始接收,将vk1主峰部分全部接收入减压蒸馏釜备用;4)进第二针(1)将一份样品溶液用流动相泵抽入色谱柱,流速5l/min;用甲醇和乙酸乙酯配制的流动相清洗样品瓶,清洗液一并抽入色谱柱以清洗进样管路,清洗标准为泵头前段透明管路中无明显颜色即可;(2)样品进入柱后,按照设定好的方法进行洗脱,当vk1峰升起即可开始接收,将vk1主峰部分全部接收入减压蒸馏釜备用;5)冲柱用乙酸乙酯冲洗柱子至检测基线平衡,流速10l/min,冲洗约10min;再用甲醇和乙酸乙酯配制的流动相冲洗柱子,流速10l/min,时间约20min。6)进第三针(1)将一份样品溶液用流动相泵抽入色谱柱,流速5l/min;用甲醇和乙酸乙酯配制的流动相清洗样品瓶,清洗液一并抽入色谱柱以清洗进样管路,清洗标准为泵头前段透明管路中无明显颜色即可;(2)样品进入柱后,按照设定好的方法进行洗脱,当vk1峰升起即可开始接收,将vk1主峰部分全部接收入减压蒸馏釜备用;7)进第四针(1)将一份样品溶液用流动相泵抽入色谱柱,流速5l/min;用甲醇和乙酸乙酯配制的流动相清洗样品瓶,清洗液一并抽入色谱柱以清洗进样管路,清洗标准为泵头前段透明管路中无明显颜色即可;(2)样品进入柱后,按照设定好的方法进行洗脱,当vk1峰升起即可开始接收,将vk1主峰部分全部接收入减压蒸馏釜备用;8)冲柱与封柱用乙酸乙酯冲洗柱子至检测基线平衡,流速10l/min,冲洗约10min;再用甲醇冲洗柱子,流速10l/min,时间约20min。9)减压蒸除溶剂将所收集的洗脱液减压浓缩,蒸出溶剂,加热从30℃逐渐升温至70℃,减压真空≥0.095mpa,抽蒸5~10小时,检测折光率1.520~1.528之间,出料,得到vk1半成品,供正相层析精制。二、工序二:正相柱层析1、物料和设备信息1-1、物料配比1-2设备信息设备名称规格搪玻璃浓缩釜2000l搪玻璃浓缩釜3000l层析柱20cm*200cm匀浆罐300l2、操作过程1)称取洗脱液称取223.30kg正己烷加入到流动相釜中备用;2)装柱称取计量的硅胶,用正己烷淹没,浸泡(加以搅拌)60分钟。然后将浸泡过的硅胶用洗脱液按湿法装柱操作,装入层析柱中;再在硅胶上层铺垫一层活性炭0.39kg(活性炭用正己烷浸泡后上柱,防止粉尘飞扬,浸泡所用正己烷从流动相釜中放出),保持硅胶及活性炭被洗脱液淹没、备用(后续操作避日光及白炽灯光)。2)装料将称取的vk1半成品加入19.80倍质量的正己烷(vk1半成品3.76kg,正己烷74.46kg),室温搅拌30分钟,稀释溶解均匀。柱层析采用下行法层析:对已装填完毕的硅胶层析柱,打开底部料液出口,将柱中洗脱液放出至柱顶部液体和硅胶平,然后从柱上端泵入已稀释溶解的粗品料液,并将柱中洗脱液从下部放出,使料液逐渐进入柱中,适当加压,使上料时间大约在1小时左右。3)洗脱待料液全部进入硅胶柱中后(料液液面与活性炭层基本持平),再从柱上部加入正己烷,从柱下部放出洗脱液,进行洗脱。整个洗脱过程,层析柱上部加入的洗脱液不能中断、不能使活性炭层露出液面,控制压力(约0.03mpa),使流出速度大约在2~3kg/min。前洗脱无色溶液返回流动相釜继续使用,待洗脱出黄色料液,开始收集,收集时间约2~3小时;将所收集的层析所得产品料液减压浓缩,真空控制在0.090~0.095mpa,控温50℃,蒸干洗脱液溶剂,至基本无冷凝液滴出。再采用高真空,控0.0980~0.1000mpa,控温70℃,抽拉4~6小试。至料液检测折光1.525~1.528,得vk1合格产品,放入铝罐待qc收件后入库。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1