吡咯嘧啶五元氮杂环化合物的晶型的制作方法
吡咯嘧啶五元氮杂环化合物的晶型
1.本技术要求于2020年10月30日向国家知识产权局递交的申请号为202011199567.8、发明名称为“吡咯嘧啶五元氮杂环化合物的晶型”的在先申请的优先权,该申请通过全文引用的方式结合于本技术中。
技术领域
2.本发明属于药物晶型技术领域,具体涉及一种吡咯嘧啶五元氮杂环化合物的新晶型,其制备方法和应用。
背景技术:
3.jak(janus kinase)激酶是一个细胞内非受体酪氨酸激酶家族,介导细胞因子产生的信号,并通过jak-stat信号通路传递下去。目前有四种已知的jak家族成员:jak激酶1(jak1)、jak激酶2(jak2)、jak激酶3(jak3)和酪氨酸激酶2(tyrosine kinase,tyk2)。jak-依赖性细胞因子参与多种炎症和自身免疫性疾病的发病过程,jak抑制剂或可广泛用于治疗各种炎性疾病。
4.专利cn201710055590.1公开了式(i)所示化合物,该化合物为新型jak激酶抑制剂,对jak2具有高效选择性,在作为类风湿性关节炎、真性红细胞增多症、银屑病、原发性血小板增多症、骨髓纤维化等jak激酶相关疾病的治疗剂时,能够降低副作用,提高安全性。
[0005][0006]
根据专利cn201710055590.1所制备得到的固体化合物的引湿性较高,稳定性、溶解度与生物利用度较低,不能满足临床的药用需求。
[0007]
固体化学药物晶型不同,可造成其溶解度和稳定性不同,从而影响药物的吸收和生物利用度,并且会导致临床药效的差异。然而,目前尚无式(i)化合物的晶型的相关报导,因此,有必要对式(i)化合物进行全面系统的多晶型筛选,选择最适合开发的晶型。
技术实现要素:
[0008]
本发明的目的在于提供式(i)化合物的新晶型,新晶型的制备方法和用途;
[0009][0010]
本发明的目的可以通过下述技术方案实现:
[0011]
本发明首先提供式(i)所示化合物的晶型a,所述晶型a以2θ角度表示的x-射线粉末衍射图具有以下特征峰:5.6
±
0.2
°
、16.9
±
0.2
°
、18.4
±
0.2
°
、18.6
±
0.2
°
和28.3
±
0.2
°
。
[0012]
优选地,本发明提供的晶型a,还在2θ为11.2
±
0.2
°
、12.0
±
0.2
°
、12.9
±
0.2
°
、13.4
±
0.2
°
、14.8
±
0.2
°
、17.6
±
0.2
°
、19.3
±
0.2
°
、20.5
±
0.2
°
、21.3
±
0.2
°
、22.5
±
0.2
°
、23.0
±
0.2
°
、24.8
±
0.2
°
、25.4
±
0.2
°
、27.9
±
0.2
°
、30.0
±
0.2
°
、30.7
±
0.2
°
、33.4
±
0.2
°
、34.9
±
0.2
°
处具有特征峰。
[0013]
更优选地,本发明提供的晶型a,其x射线粉末衍射图谱基本上与图1一致。
[0014]
本发明提供的晶型a,tga和dsc结果如图2所示,样品加热至150℃失重为1.9%,在234.1℃附近开始出现吸热峰。由此可知,晶型a为无水晶型。
[0015]
本发明还涉及所述晶型a的制备方法:将式(i)化合物的磷酸盐在甲醇、水、二甲亚砜/乙酸异丙酯、二甲亚砜/甲苯或乙醇/水中结晶制得。
[0016]
本技术发明人在进行式(i)化合物的晶型研究研究时,发现式(i)化合物的溶解度极低,大多数溶剂都无法将其溶解,故无法对其进行晶型研究。而本技术发明人在对其磷酸盐进行晶型研究时,意外发现磷酸盐晶型在一定条件下会转换为式(i)化合物游离碱晶型。如在甲醇、乙醇、异丙醇,n,n-二甲基甲酰胺、水等多种体系中观察到磷酸盐晶型可歧化成游离碱晶型a。通过进一步研究发现,将式(i)化合物的磷酸盐在部分溶剂中,如甲醇、水、二甲亚砜/乙酸异丙酯、二甲亚砜/甲苯或乙醇/水中结晶可制得式(i)化合物游离碱晶型,命名为晶型a。
[0017]
使用乙醇/水结晶时,当乙醇与水的比例小于6:1时,得到式(i)化合物游离碱晶型a。
[0018]
作为优选,使用甲醇、水、二甲亚砜/乙酸异丙酯或二甲亚砜/甲苯作溶剂结晶制备式(i)化合物游离碱晶型a。
[0019]
本发明的另一个目的是提供式(i)化合物的磷酸盐晶型b,所述晶型b以2θ角度表示的x-射线粉末衍射图具有以下特征峰:15.19
±
0.2
°
、18.56
±
0.2
°
、22.89
±
0.2
°
。
[0020]
优选地,本发明提供的晶型b,还在2θ为7.63
±
0.2
°
、10.57
±
0.2
°
、13.59
±
0.2
°
、14.41
±
0.2
°
、16.22
±
0.2
°
、16.73
±
0.2
°
、18.21
±
0.2
°
、19.05
±
0.2
°
、19.47
±
0.2
°
、20.51
±
0.2
°
、21.16
±
0.2
°
、21.94
±
0.2
°
处具有特征峰。
[0021]
更优选地,本发明提供的晶型b,其x射线粉末衍射图谱基本上与图3一致。
[0022]
本发明提供的晶型b,tga和dsc结果如图4所示,样品在150℃前失重2.0%,在
216.3℃附近开始出现吸热峰。由此可知,晶型b为无水晶型。
[0023]
本发明提供了晶型b的制备方法:由式(i)化合物和磷酸在乙醇/乙腈溶剂中反应制备。
[0024]
其中,乙醇与乙腈的体积比为1:3时,可较满意地制得式(i)化合物磷酸盐的晶型b。经ic检测,制得的式(i)化合物的晶型b中po
43-离子含量为19.3%(理论值19.1%),故晶型b中1分子游离碱结合1分子磷酸。
[0025]
本技术发明人使用其他有机溶剂,如乙醇、丙酮、乙酸乙酯、甲基叔丁基醚、2-甲基四氢呋喃等作反应溶剂时,得到的固体中磷酸根离子含量与理论值有一定差别。使用体积比为3:1的乙醇/乙腈混合溶剂时,无法得到磷酸盐化合物。
[0026]
优选地,制备晶型b时,磷酸和式(i)化合物的摩尔比在1.5以上。
[0027]
本技术发明人对晶型b的歧化现象进行了研究。在乙醇溶液中,晶型b直接搅拌4天,会有部分歧化为游离碱晶型a。而将式(i)游离碱固体和1.5当量以上的磷酸在乙醇中搅拌3天,仅生成磷酸盐晶型b。故在制备磷酸盐晶型b时,需加入1.5当量以上的磷酸。
[0028]
本发明还提供了一种式(i)化合物的晶型c。其x射线粉末衍射图谱基本上与图5一致。晶型c可由晶型b在dmf和dcm的混合溶剂中结晶制得,但在室温下放置过夜后转变为游离碱晶型a和磷酸盐晶型b的混合晶型。
[0029]
本发明的另一个目的是提供一种药用组合物,包含有效治疗量的晶型a或晶型b以及药学上可接受的载体、稀释剂或赋形剂。
[0030]
本发明提供的式(i)化合物的游离碱新晶型a、磷酸盐新晶型b或二者的混合物可用于治疗自身免疫疾病的药物的制备,特别是用于治疗类风湿性关节炎药物制剂的用途。
[0031]
综上,目前尚无专利或文献报导式(i)化合物的晶型,式(i)化合物溶解度很低,难以进行晶型研究,本发明的发明人突破了这一难题,找到了适合开发的两种新晶型。
[0032]
本发明提供的晶型稳定性好,工艺提纯效果显著。能很好地避免药物储存以及开发过程中发生转晶,从而避免生物利用度以及药效的改变。
[0033]
本发明提供的晶型a和b无引湿性或引湿性低,满足生物利用度和药效要求,在制备工艺中无需特殊的干燥条件,简化了药物的制备工艺和后处理工艺,且不易受湿度影响,对储存条件要求不苛刻,便于长期贮存,大大降低了物料储存以及质量控制方面的成本,具有很强的经济价值。
[0034]
综上,本发明提供的晶型a和b较现有技术具有显著的进步,尤其是相比于专利cn201710055590.1实施例1所制备得到固体化合物相比具有更低的吸湿性,更高的稳定性、溶解度与生物利用度,为以后制剂研究提供了支持,从而满足不同临床用药需求。
附图说明
[0035]
图1是本发明晶型a的x-射线粉末衍射(xrd)图;
[0036]
图2是本发明晶型a的热重分析(tga)及差示扫描分析(dsc)图;
[0037]
图3是本发明磷酸盐晶型b的x-射线粉末衍射(xrd)图;
[0038]
图4是本发明磷酸盐晶型b的热重分析(tga)及差示扫描分析(dsc)图;
[0039]
图5是本发明晶型c的x-射线粉末衍射(xrd)图;
[0040]
图6是本发明游离碱晶型a的动态水分吸附(dvs)图;
[0041]
图7是本发明游离碱晶型a的动态水分吸附(dvs)测试前后的xrpd叠图;
[0042]
图8是本发明磷酸盐晶型b的动态水分吸附(dvs)图;
[0043]
图9是本发明磷酸盐晶型b的动态水分吸附(dvs)测试前后的xrpd叠图。
具体实施方式
[0044]
为了更好地理解本发明的内容,下面结合具体实施例对本发明的技术方案做进一步的说明,但具体的实施方式并不意味着对本发明有任何限制。
[0045]
检测仪器及方法:
[0046]
x-射线粉末衍射(xrd)所使用的仪器为bruker d8 advance diffractometer,配置有θ-2θ测角仪、mo单色仪、lynxeye探测器。采集软件是diffrac plus xrdcommander,分析软件是mdi jade 5.0。仪器在使用前用仪器自带的标准品(一般为刚玉)校准。检测条件为:2θ扫描角度范围3~40
°
,步长0.013
°
,连续扫描7分钟。检测过程:采用铜靶波长为1.54nm的ka x-射线,在45kv和40ma的操作条件下,样品在室温条件下测试,把需要检测的样品放在有机玻片上。除非特别说明,样品在检测前未经研磨。
[0047]
热重分析(tga):仪器型号:ta/tga q500,吹扫气:氮气40ml/min,升温速度:10℃/min,温度范围:5℃~350℃。
[0048]
差示扫描量热分析仪(dsc):仪器型号:ta/dsc q200,吹扫气:氮气40ml/min,升温速度:10℃/min,温度范围:20℃~250℃。
[0049]
动态水分吸附仪(dvs):吹扫气:氮气200ml/min,升温速度:10℃/min,dm/dt:0.002%/分钟,rh范围:0%rh-95%rh-0%rh,rh梯度:5%。
[0050]
实施例所用固体参照专利cn201710055590.1实施例1制得。
[0051]
实施例1
[0052]
反应瓶中加入游离碱固体(53g,0.1328mol)溶于乙腈(2.4l)和乙醇(800ml)的溶液,加入磷酸(20g,0.2mol),80℃搅拌过夜。反应体系冷却至室温,有固体析出,过滤,得磷酸盐晶型b固体(50.3g,收率:76%,po
43-含量:19.3%)。
[0053]
ms(m/s):400[m+h]
+
;
[0054]1h-nmr(400mhz,dmso-d6):δ1.17(t,j=7.4hz,3h),2.14(m,j=8.0hz,2h),2.83(d,j=10hz,2h),2.96(t,j=10.6hz,2h),3.05(m,j=7.3hz,2h),3.29(s,2h),3.56(m,j=4.2hz,2h),7.10(m,j=1.7hz,1h),7.63(t,j=2.0hz,1h),8.45(s,1h),8.71(s,1h),8.82(s,1h),12.15(s,1h)。
[0055]
采用相似的方法,使用乙腈作溶剂时,得到的固体中po
43-含量为23.1%,使用乙醇或异丙醇作溶剂时,得到的固体中po
43-含量约为22.1%。
[0056]
实施例2
[0057]
称取约15mg磷酸盐晶型b于20毫升小瓶内,加入二甲亚砜溶解至澄清,向该澄清溶液中逐滴加入异丙醇,边滴加边搅拌至有固体析出。收集固体并进行xrpd测试,得游离碱晶型a。xrpd谱图见图1。
[0058]
采用相同的实验方法,使用二甲亚砜/甲基异丁基酮、二甲亚砜/乙酸乙酯、n/n-二甲基甲酰胺/甲苯、n/n-二甲基甲酰胺/乙酸异丙酯、n/n-二甲基甲酰胺/1,4二氧六环、n/n-二甲基甲酰胺/甲基叔丁基醚、乙醇/二氯甲烷(1:2)/甲苯、乙醇/二氯甲烷(1:2)/正庚烷等
作结晶溶剂时,得到磷酸盐晶型b。xrpd谱图见图3。
[0059]
实施例3
[0060]
称量约10mg磷酸盐晶型b至3毫升小瓶中,加入约2.0-2.4ml甲醇配制成澄清溶液,或用尼龙滤膜(0.45μm)过滤后得到澄清液,置于室温下用封口膜密封,刺2~4个小孔后放置自然挥发,收集所得固体并进行xrpd测试,得游离碱晶型a。
[0061]
采用相同的实验方法,使用乙醇/二氯甲烷(v/v=1:2)、乙醇/氯仿(v/v=1:3)作结晶溶剂时,得到磷酸盐晶型b。
[0062]
实施例4
[0063]
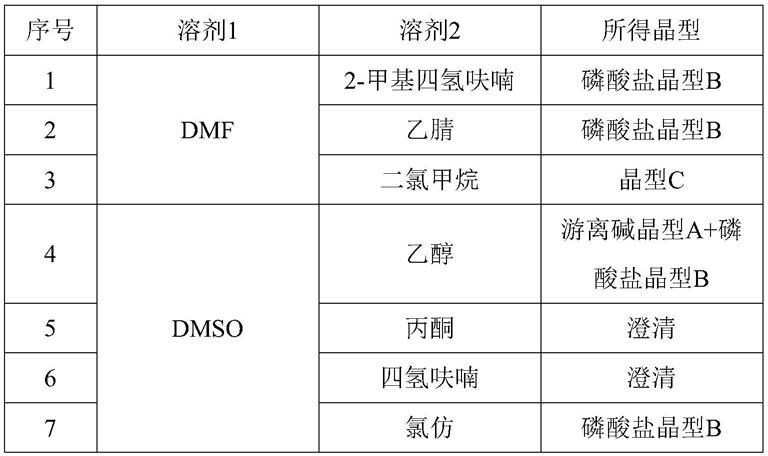
称取约15mg每份的磷酸盐晶型b溶于一定量的溶剂1中,过滤取滤液于3毫升小瓶中,另取20毫升的小瓶并向其中加入约3毫升的溶剂2,将3毫升小瓶敞口置于20毫升小瓶中,密封并于室温下静置。当观察到有固体析出时,则取出固体进行xrpd测试。试验结果如表1所示。
[0064]
表1
[0065][0066]
晶型c在室温下放置过夜后转变为游离碱晶型a和磷酸盐晶型b的混合物,xrpd对比谱图见图5。
[0067]
实施例5
[0068]
称取约20mg磷酸盐晶型b于3毫升小瓶中,加入1.0ml二甲亚砜/甲苯(v/v=1:5)的混合溶剂,在50℃下搅拌1小时后取清液过滤(聚四氟乙烯滤膜,0.45μm),将所得滤液以0.1℃/min的速度从50℃降温至5℃并在5℃恒温。若样品澄清,则转至室温挥发。收集析出的固体并进行xrpd测试,得到游离碱晶型a。
[0069]
采用相同的实验方法,使用乙醇/氯仿(v/v=1:3)作结晶溶剂时,得到磷酸盐晶型b。使用n,n-二甲基甲酰胺/甲基异丁基酮(v/v=1:5)或甲醇/水(v/v=3:1)作结晶溶剂时,体系澄清,无固体析出。
[0070]
实施例6
[0071]
称量约15mg每份的磷酸盐晶型b至1.5毫升玻璃小瓶中,分别加入0.5毫升表2中所列的溶剂,得到的悬浮液于室温下搅拌约4天后,离心收集固体并进行xrpd测试。试验结果
见表2。
[0072]
表2
[0073][0074][0075]
实施例7
[0076]
称量约20mg每份的磷酸盐晶型b样品至hplc玻璃小瓶中,加入0.5ml甲醇,得到的悬浮液在50℃下搅拌约4天后,离心收集固体并进行xrpd测试,得到游离碱晶型a。
[0077]
采用相同的实验方法,使用丙酮、乙酸乙酯、氯仿、四氢呋喃、2-甲基四氢呋喃或乙腈作结晶溶剂时,得到磷酸盐晶型b。使用乙醇作溶剂时,得到游离碱晶型a和磷酸盐晶型b的混晶。
[0078]
实施例8
[0079]
为评估磷酸盐晶型b在etoh中的歧化风险,在室温条件下考察不同酸碱摩尔比时磷酸盐晶型b的稳定性。结果如下表3。
[0080]
表3
[0081]
[0082][0083]
实施例9
[0084]
为评估游离碱晶型a和磷酸盐晶型b在不同湿度条件下的稳定性,将样品预先在0%rh条件下干燥去除可能的吸附溶剂或水,在25℃恒温条件下进行dvs测试。
[0085]
当湿度达到80%时,游离碱晶型a水分吸附量为0.015%,表明游离碱晶型a几乎无引湿性,且dvs测试前后晶型不变,如附图6和附图7所示。磷酸盐晶型b水分吸附量为1.17%,表明磷酸盐晶型b具有轻微引湿性,xrpd结果显示在dvs测试前后样品晶型不变,如附图8和附图9所示。
[0086]
实施例10
[0087]
考察不同条件下游离碱晶型a和磷酸盐晶型b的固态稳定性,条件及结果如下表4所示。
[0088][0089]
结果表明,游离碱晶型a和磷酸盐晶型b样品的晶型和纯度均没有发生明显变化,由此可知,游离碱晶型a和磷酸盐晶型b具有较好的物理和化学稳定性。
[0090]
实施例11
[0091]
游离碱晶型a的生物利用度测试
[0092]
实验动物:雄性cd-1小鼠6只。
[0093]
给药方案:静脉注射剂量为1.0mg/kg,给药容量为1ml/kg;口服剂量为5mg/kg。
[0094]
静脉注射药物配制:称取适量ob756,用5%dmac、5%solutol和90%saline来溶解,浓度为0.2mg/ml。
[0095]
口服用药配制:称取适量ob756,用0.5%mc和0.4%tween80配制成均匀悬浮液,浓度为0.5mg/ml。
[0096]
用lc-ms测定小鼠血液样品,结果显示游离碱晶型a在cd-1小鼠中的平均生物利用度(f)为28.8%。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1