苯乙炔类液晶材料的制备方法与流程
本发明涉及液晶材料领域,具体而言,涉及一种苯乙炔类液晶材料的制备方法。
背景技术:
立体显示是显示领域的发展方向,尤其是裸眼3D显示无需佩戴眼镜,将会是3D显示的主流趋势。
裸眼3D显示装置中,需要一种分光的器件将显示器中有区别的像素信息准确的传送到人的左、右眼,常用的分光器件有透镜、狭缝等。透镜可以大体分为两种,一种是具有透镜常规的物理形状,比如曲面结构,常使用可聚合各向异性的光学材料通过UV聚合或热聚合等方法制备,但是不适合大规模量产,且良率较低;另一种为液晶透镜,将液晶材料夹在两层电极基板中间,利用液晶材料的电响应特性,施加电压改变液晶层的光学性质,通过电场梯度作用可实现透镜的效果,且液晶透镜可以使用现有液晶显示屏的生产线来进行生产,利于大规模量产和推广。
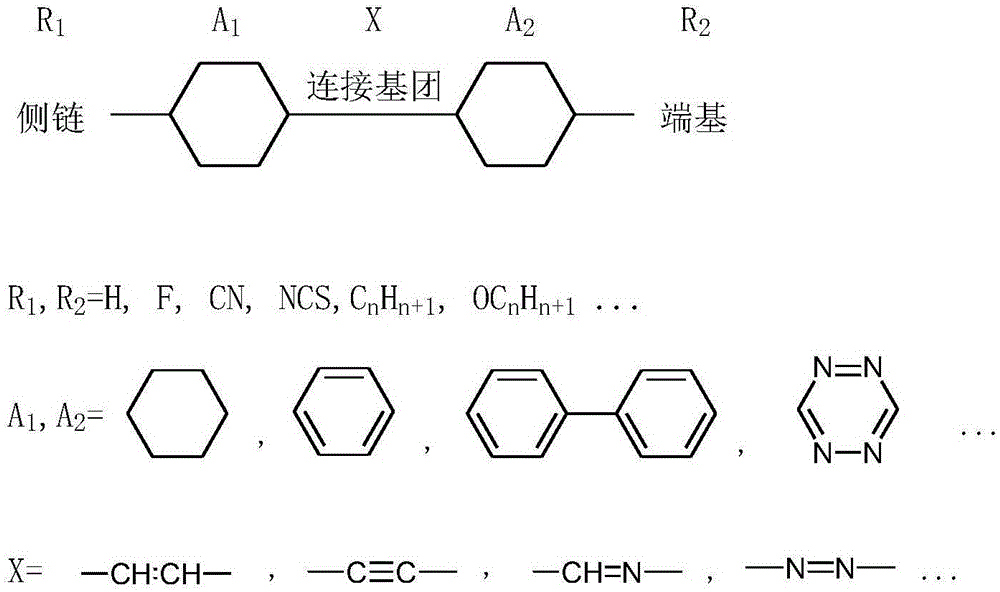
液晶透镜中常用到的高双折射率液晶材料一般为一种小分子化合物的混合物,图1为高双折射率液晶材料小分子的常见分子结构,两个带侧链(R1)或端基(R2)的环结构(A1,A2)由连接基团(X)连接起来。
图2为高双折射率液晶材料中一类常用的苯乙炔类分子结构,由于其中间连接基团为乙炔,这类化学结构的液晶单体能使液晶材料的粘度显著降低,是高双折射率液晶材料中非常重要的一类。因此被广泛应用于液晶材料中,也是研究最多的一类液晶材料。
对于图2的苯乙炔类液晶分子结构的合成,现有技术中一般采用sonogashira反应制备此类单体液晶,如图3所示。
sonogashira反应在此类结构合成中具有一些不可替代的优势,如反应产 率较高,条件相对温和,反应步骤简单等。但是,sonogashira反应也有一些不可避免的缺点,如催化剂钯不易去除,残留的金属钯将会较大地降低液晶材料的电阻率,并影响其在显示器件中的响应电压;又如反应底物炔在sonogashira反应条件下会产生自身偶合的副产物,使后处理纯化变得复杂。催化剂钯在使用柱层析色谱法纯化时,容易与产物一起被冲刷出来,因此产物很容易残留钯;而反应底物的自身偶合副产物会因为反应体系中的水和氧残余量,底物本身活性等条件不同而产生的量也不同,现实操作中很难通过控制这些条件来避免此类自身偶合副产物的产生。特别是在制备氰基(CN)与异硫腈基(NCS)类液晶单体时,由于自身偶合副产物与最终产物CN,NCS类液晶单体的极性非常接近,自身偶合产物与最终产物混合在一起时,用柱层析色谱法很难将他们分离,导致最终产物纯化变得复杂,因此在制备最终反应产物CN,NCS类液晶单体步骤前,必须把自身偶合副产物除去,保证最终产物的纯度。
目前,用于sonogashira后处理的常用纯化方法是柱层析色谱法,即利用产物与副产物的不同极性来对产物和副产物进行分离,从而达到纯化目的,但由于柱层析色谱法在制备量变大时,所需花费的准备时间,分离时间和洗脱溶剂大大增加,并且相对应的柱层析柱子也要加大,非常不利于工业化的大量生产。
因此需提供一个简便有效的方法来制备苯乙炔类液晶材料,简化产物提纯方法,并且保证产率和纯度较高,便于大量生产。
技术实现要素:
为了简化苯乙炔类液晶材料的提纯方法,本发明提出一种苯乙炔类液晶材料的制备方法,所述苯乙炔类液晶材料如式I所示:
(I),
所述苯乙炔类液晶材料的制备包括如下步骤:
1)合成:在第一有机溶剂里,将式II的化合物(II)、式III的化合物(III)和碱,在催化剂的作用下加热发生sonogashira反应:
其中,R1为-F或-NH2,R2为具有3-7个碳原子的烷基、具有3-7个碳原子的不饱和烃基或具有3-7个碳原子的烷氧基,L1和L2分别为H或F,m为0或1,X为-Cl、-Br或-I,为或
2)提纯:上述反应完成后,用索式提取器对步骤1)反应产物进行提纯。
进一步的,所述步骤2)的所述提纯包括以下步骤:
a.将所述反应液倒入冰水,搅拌并滴加盐酸至弱碱性;
b.用第二有机溶剂萃取所述步骤a所得的液体,收集有机相,先用盐水洗所述有机相,接着用无水硫酸钠干燥所述有机相,最后,旋转蒸发干所述有机相得褐色固体;
c.在索氏提取器的提取瓶中加入石油醚,提取管加入所述步骤b所得的褐色固体,加热回流1~2小时后,将提取瓶的石油醚冷却至室温,产物固体析出,利用抽滤装置过滤得到提纯产物。
更进一步的,其特征在于,所述第二有机溶剂选自二氯甲烷、乙酸乙酯和乙醚中的一种。
进一步的,所述步骤1)的反应在惰性气氛中进行。
进一步的,所述步骤1)中,所述第一溶剂选自四氢呋喃,N,N-二甲基甲酰胺,二甲基亚砜和1,4-二氧六环中的一种。
进一步的,所述步骤1)中,所述碱选自N,N-二乙基乙二胺,N,N-二 异丙基乙二胺和三乙胺中的一种。
进一步的,所述步骤1)中,所述催化剂包括钯催化剂。
更进一步的,所述催化剂选自Pd(Pph3)4,Pb(dppf)Cl2和Pd(Pph3)2Cl2中的一种。
更进一步的,所述催化剂还包括碘化亚铜,氯化锌或正丁基锂中的一种。
进一步的,所述步骤1)的反应时间为4~8小时。
进一步的,所述制备方法进一步包括步骤3):在第三有机溶剂里,加入步骤2)所得的提纯产物与CSCl2,所述步骤2)的提纯产物与CSCl2发生如下反应:
其中,R1为-NH2,R2为具有3-7个碳原子的烷基、具有3-7个碳原子的不饱和烃基或具有3-7个碳原子的烷氧基,L1和L2分别为H或F,m为0或1,
为或
更进一步的,所述第三有机溶剂选自氯仿和二氯甲烷。
进一步的,所述制备方法进一步包括步骤3):加入步骤2)所得的提纯产物与亚硝酸钠水溶液,并向反应液滴加溶于氨与氯化铵缓冲溶液的镍氰化钾,所述步骤2)的提纯产物与镍氰化钾发生如下反应:
其中,R1为-NH2,R2为具有3-7个碳原子的烷基、具有3-7个碳原子的不饱和烃基或具有3-7个碳原子的烷氧基,L1和L2分别为H或F,m为0或1,
为或
更进一步的,所述制备方法具体包括:
a:在反应瓶中加入步骤2)所得的提纯产物和稀盐酸,并将反应置于冰浴中,缓慢滴加亚硝酸钠水溶液,在冰浴条件下反应1~4小时;
b:上述反应完成后,用碳酸钠溶解将上述反应液调至中性,并向反应液滴加溶于氨与氯化铵缓冲溶液的镍氰化钾,然后将反应升至60℃。
附图说明
图1为高双折射率液晶材料常见分子结构图。
图2为高双折射率液晶材料苯乙炔类分子结构图。
图3苯乙炔类分子的sonogashira合成方法。
图4为实施例1利用索氏提取法提纯产物过程图。
具体实施方式
为了能够更清楚地理解本发明的上述目的、特征和优点,下面结合附图和具体实施方式对本发明进行进一步的详细描述。需要说明的是,在不冲突的情况下,本申请的实施例及实施例中的特征可以相互组合。
在下面的描述中阐述了很多具体细节以便于充分理解本发明,但是,本发明还可以采用其他不同于在此描述的其他方式来实施,因此,本发明并不限于下面公开的具体实施例的限制。
实施例1
本实施例为的制备:
步骤1),合成目标产物:
搭建回流反应装置,向反应瓶内放入搅拌子,加入2,6-二氟-4-溴苯胺、催化剂Pd(Pph3)4、碘化亚铜;密封反应系统,将密封系统里的空气置换成氩气;注入三乙胺、四氢呋喃和4-丙基苯乙炔,搅拌并慢慢加热回流,确保系统在氩气保护下反应。4小时后,用TLC检测反应程度。
步骤2),用索式提取器对步骤1)的产物进行提纯:
反应完毕后,将反应液倒入冰水,滴加稀盐酸并搅拌,滴至弱碱性,pH为8~9;用二氯甲烷萃取上述液体,收集有机相,所得有机相用盐水洗两次后,接着用无水硫酸钠或无水硫酸镁干燥,最后,旋转蒸发干有机相得褐色固体,此时褐色固体为sonogashira反应的粗产品,包含目标产物、自身耦合副产物以及其他副产物。
搭建好提取装置,在索氏提取器的提取瓶内倒入石油醚;将上述褐色固体装于滤纸筒里面,将滤纸筒放置在索氏提取器的提取管;加热回流1~2小时,将提取瓶的石油醚冷却至室温或更低,产物固体析出,利用抽滤装置过滤,获得纯度95%以上产物。
索氏提取法与柱层析色谱法对比:
本发明利用索式提取器对步骤1)的产物粗产品进行纯化处理,产物粗产品包括目标产物、自身耦合副产物以及其他副产物。由于产物,自身偶合副产物及其他副产物在石油醚里的不同溶解性,溶解性为:自身偶合副产物(易溶)>产物(部分溶解)>其他副产物(难溶),且金属钯在石油醚里难溶。
提纯原理如图4所示,随着石油醚的加热回流,由于自身偶合副产物易溶于石油醚中,首先随石油醚一起流入提取瓶;部分溶解的目标产物也随着抽提的次数增加,全部被抽提到提取瓶,而难溶于石油醚的其他副产物(金属钯催化剂等)则留在提取管中。
待抽提完毕,冷却提取瓶,在冷却过程中,目标产物慢慢从石油醚中析出,此时只需抽滤,收集滤固,即可获得到纯度95%以上产物,若需要更纯产物,再次重结晶便可。
步骤3),制备最终产物:
搭建回流反应装置,向反应瓶内放入搅拌子,加入上述步骤2)提纯后的产物和溶剂二氯甲烷或氯仿,搅拌溶解,并将反应瓶置于冰浴中,待冷至冰浴温度,缓慢滴加CSCl2,滴加完毕,移除冰浴,反应液慢慢升至室温后,开始对反应加热回流。12个小时后,用TLC检测反应程度;反应完毕后,加入碳酸钠溶液,使过量的CSCl2消耗完,然后分离出有机相,所得有机相用盐水洗两次后,接着用无水硫酸钠或无水硫酸镁干燥,最后,旋转蒸发干有机相得红褐色油状物,利用柱层析色谱法可分离出纯度为98%以上的最终产物。
此外,步骤2)对产物的提纯,对于步骤3)的NCS类最终产物(或CN类最终产物)提纯比较关键。由于步骤1)反应过程产生的自身偶合副产物与步骤3)的最终产物极性非常接近,用柱层析色谱法提纯最终产物时,若不对自身偶合副产物提前处理,自身偶合副产物与最终产物很难分离,必须经过多次分离,这必然导致产率降低,甚至部分结构经多次分离也无法分离干净,导致纯度无法提升。因此必须提前除去自身偶合副产物,利用以上索氏提取的方法便可巧妙地解决自身偶合副产物对CN类或NCS类苯乙炔液晶单体最终产物提纯的影响。
本发明提供的苯乙炔类液晶材料的制备方法,操作方便,仪器装置简单,只需索氏提取装置和使用抽滤装置即可;节省溶剂,利用索氏提取器,相比柱层析色谱法只需较少的溶剂即可完成抽提;提纯损失少,产率在一般70%以上,比柱层析色谱法的产率高;获取的产物纯度高,普遍在95%以上;由于仪器,步骤简单,所需溶剂少,耗费时间少等优势,因此,此方法在工业化的大量生产中具有可行性。
实施例2
本实施例为的制备:
步骤1),合成目标产物:
搭建回流反应装置,向反应瓶内放入搅拌子,加入2-氟-4-溴苯胺、催 化剂Pb(dppf)Cl2、氯化锌;密封反应系统,将密封系统里的空气置换成氩气;注入N,N-二异丙基乙二胺、二甲基亚砜和4-戊基苯乙炔,搅拌并慢慢加热回流,确保系统在氩气保护下反应。4小时后,用TLC检测反应程度。
步骤2),用索式提取器对步骤1)的产物进行提纯:
反应完毕后,将反应液倒入冰水,滴加稀盐酸并搅拌,滴至弱碱性,pH为8~9;用二氯甲烷萃取上述液体,收集有机相,所得有机相用盐水洗两次后,接着用无水硫酸钠或无水硫酸镁干燥,最后,旋转蒸发干有机相得褐色固体,此时褐色固体为sonogashira反应的粗产品,包含目标产物、自身耦合副产物以及其他副产物。搭建好提取装置,在索氏提取器的提取瓶内倒入石油醚;将上述褐色固体装于滤纸筒里面,将滤纸筒放置在索氏提取器的提取管;加热回流1-2小时,将提取瓶的石油醚冷却至室温或更低,产物固体析出,利用抽滤装置过滤,获得纯度95%以上产物。
索氏提取法与柱层析色谱法对比:
步骤3),制备最终产物:
搭建回流反应装置,向反应瓶内放入搅拌子,加入上述步骤2)提纯后的产物和溶剂二氯甲烷或氯仿,搅拌溶解,并将反应瓶置于冰浴中,待冷至冰浴温度,缓慢滴加CSCl2,滴加完毕,移除冰浴,反应液慢慢升至室温后,开始对反应加热回流。12个小时后,用TLC检测反应程度;反应完毕后,加入碳酸钠溶液,使过量的CSCl2消耗完,然后分离出有机相,所得有机相用盐水洗两次后,接着用无水硫酸钠或无水硫酸镁干燥,最后,旋转蒸发干有机相得红褐色油状物,利用柱层析色谱法可分离出纯度为98%以上的最终产物。
实施例3
本实施例为的制备:
步骤1),制备最终产物:
搭建回流反应装置,向反应瓶内放入搅拌子,加入4-丙基环己基乙炔、2-氟-4-溴苯胺、催化剂Pd(Pph3)2Cl2、正丁基锂;将密封系统里的空气置换成氩气;注入N,N-二乙基乙二胺、1,4-二氧六环,搅拌并慢慢加热回流,确保系统在氩气保护下反应。4小时后,用TLC检测反应程度。
步骤2),用索式提取器对步骤1)的产物进行提纯:
反应完毕后,将反应液倒入冰水,滴加稀盐酸并搅拌,滴至弱碱性,pH为8~9;用二氯甲烷萃取上述液体,收集有机相,所得有机相用盐水洗两次后,接着用无水硫酸钠或无水硫酸镁干燥,最后,旋转蒸发干有机相得褐色固体,此时褐色固体为sonogashira反应的粗产品,包含目标产物、自身耦合副产物以及其他副产物。搭建好提取装置,在索氏提取器的提取瓶内倒入石油醚;将上述褐色固体装于滤纸筒里面,将滤纸筒放置在索氏提取器的提取管;加热回流1-2小时,将提取瓶的石油醚冷却至室温或更低,产物固体析出,利用抽滤装置过滤,获得纯度95%以上产物。
索氏提取法与柱层析色谱法对比:
本发明提供的苯乙炔类液晶材料的制备方法,操作方便,仪器装置简单,只需索氏提取装置和使用抽滤装置即可;节省溶剂,利用索氏提取器,相比柱层析色谱法只需较少的溶剂即可完成抽提;提纯损失少,产率在一般70%以上,比柱层析色谱法的产率高;获取的产物纯度高,普遍在95%以上;由于仪器,步骤简单,所需溶剂少,耗费时间少等优势,因此,此方法在工业化的大量生产中具有可行性。
实施例4
本实施例为的制备:
步骤1),制备目标产物:
搭建回流反应装置,向反应瓶内放入搅拌子,加入4-庚基环己基乙炔、4-溴苯胺、催化剂Pb(dppf)Cl2、碘化亚铜;将密封系统里的空气置换成氩气;注入三乙胺、四氢呋喃,搅拌并慢慢加热回流,确保系统在氩气保护下反应。5小时后,用TLC检测反应程度。
步骤2),用索式提取器对步骤1)粗产品进行提纯:
反应完毕后,将反应液倒入冰水,滴加稀盐酸并搅拌,滴至弱碱性,pH为8~9;用二氯甲烷萃取上述液体,收集有机相,所得有机相用盐水洗两次后,接着用无水硫酸钠或无水硫酸镁干燥,最后,旋转蒸发干有机相得褐色固体,此时褐色固体为sonogashira反应的粗产品,包含目标产物、自身耦合副产物以及其他副产物。搭建好提取装置,在索氏提取器的提取瓶内倒入石油醚;将上述褐色固体装于滤纸筒里面,将滤纸筒放置在索氏提取器的提取管;加热回流1-2小时,将提取瓶的石油醚冷却至室温或更低,产物固体析出,利用抽滤装置过滤,获得纯度95%以上产物。
索氏提取法与柱层析色谱法对比:
步骤3),制备最终产物:
搭建回流反应装置,向反应瓶内放入搅拌子,加入上述步骤2)提纯后的产物和溶剂二氯甲烷或氯仿,搅拌溶解,并将反应瓶置于冰浴中,待冷至冰浴温度,缓慢滴加CSCl2,滴加完毕,移除冰浴,反应液慢慢升至 室温后,开始对反应加热回流。12个小时后,用TLC检测反应程度;反应完毕后,加入碳酸钠溶液,使过量的CSCl2消耗完,然后分离出有机相,所得有机相用盐水洗两次后,接着用无水硫酸钠或无水硫酸镁干燥,最后,旋转蒸发干有机相得红褐色油状物,利用柱层析色谱法可分离出纯度为99%以上的最终产物。
实施例5
本实施例为的制备:
步骤1),制备目标产物:
搭建回流反应装置,向反应瓶内放入搅拌子,加入2-氟-4-溴苯胺、催化剂Pd(Pph3)2Cl2和碘化亚铜;将密封系统里的空气置换成氩气;注入三乙胺、N,N-二甲基甲酰胺,搅拌并慢慢加热回流,确保系统在氩气保护下反应。7小时后,用TLC检测反应程度。
步骤2),用索式提取器对步骤1)粗产品进行提纯:
反应完毕后,将反应液倒入冰水,滴加稀盐酸并搅拌,滴至弱碱性,pH为8~9;用二氯甲烷萃取上述液体,收集有机相,所得有机相用盐水洗两次后,接着用无水硫酸钠或无水硫酸镁干燥,最后,旋转蒸发干有机相得褐色固体,此时褐色固体为sonogashira反应的粗产品,包含目标产物、自身耦合副产物以及其他副产物。搭建好提取装置,在索氏提取器的提取瓶内倒入石油醚;将上述褐色固体装于滤纸筒里面,将滤纸筒放置在索氏提取器的提取管;加热回流1-2小时,将提取瓶的石油醚冷却至室温或更低,产物固体析出,利用抽滤装置过滤,获得纯度95%以上产物。
索氏提取法与柱层析色谱法对比:
步骤3),制备最终产物:
反应瓶内放入搅拌子,加入上述步骤2)提纯后的产物和稀盐酸,并将反应置于冰浴中,缓慢滴加亚硝酸钠水溶液;在冰浴条件下反应2小时候后,用碳酸钠溶解将上述反应液调至中性,并向反应液滴加溶于氨与氯化铵缓冲溶液的镍氰化钾,然后将反应升至60℃,反应15分钟,对反应液进行抽滤并用少量水洗滤固,收集滤液,用二氯甲烷对滤液经行萃取,收集有机相,所得有机相用盐水洗两次后,接着用无水硫酸镁干燥,最后,旋转蒸发干有机相得淡黄色固体,利用柱层析色谱法可分离出纯度为99%以上的最终产物。
实施例6
本实施例为的制备:
步骤1),制备目标产物:
搭建回流反应装置,向反应瓶内放入搅拌子,加入4-丙氧基环己基乙炔、2,6-二氟-4-溴苯胺、催化剂Pd(Pph3)2Cl2、碘化亚铜;将密封系统里的空气置换成氩气;注入三乙胺、四氢呋喃,搅拌并慢慢加热回流,确保系统在氩气保护下反应。5小时后,用TLC检测反应程度。
步骤2),用索式提取器对步骤1)粗产品进行提纯:
反应完毕后,将反应液倒入冰水,滴加稀盐酸并搅拌,滴至弱碱性;用二氯甲烷萃取上述液体,收集有机相,所得有机相用盐水洗两次后,接着用无水硫酸钠或无水硫酸镁干燥,最后,旋转蒸发干有机相得褐色固体,此时褐色固体为sonogashira反应的粗产品,包含目标产物、自身耦合副产 物以及其他副产物。搭建好提取装置,在索氏提取器的提取瓶内倒入石油醚;将上述褐色固体装于滤纸筒里面,将滤纸筒放置在索氏提取器的提取管;加热回流1-2小时,将提取瓶的石油醚冷却至室温或更低,产物固体析出,利用抽滤装置过滤,获得纯度98%以上产物。
索氏提取法与柱层析色谱法对比:
步骤3),制备最终产物:
反应瓶内放入搅拌子,加入上述步骤2)提纯后的产物和稀盐酸,并将反应置于冰浴中,缓慢滴加亚硝酸钠水溶液;在冰浴条件下反应2小时候后,用碳酸钠溶解将上述反应液调至中性,并向反应液滴加溶于氨与氯化铵缓冲溶液的镍氰化钾,然后将反应升至60℃,反应15分钟,对反应液进行抽滤并用少量水洗滤固,收集滤液,用二氯甲烷对滤液经行萃取,收集有机相,所得有机相用盐水洗两次后,接着用无水硫酸镁干燥,最后,旋转蒸发干有机相得淡黄色固体,利用柱层析色谱法可分离出纯度为99%以上的最终产物。
实施例7
本实施例为的制备:
步骤1),制备最终产物:
搭建回流反应装置,向反应瓶内放入搅拌子,加入4-丙基联苯乙炔、1,2,6-三氟-4-碘苯、催化剂Pd(Pph3)4和碘化亚铜;将密封系统里的空气置 换成氩气;注入三乙胺、四氢呋喃,搅拌并慢慢加热回流,确保系统在氩气保护下反应。7小时后,用TLC检测反应程度。
步骤2),用索式提取器对步骤1)粗产品进行提纯:
反应完毕后,将反应液倒入冰水,滴加稀盐酸并搅拌,滴至弱碱性;用二氯甲烷萃取上述液体,收集有机相,所得有机相用盐水洗两次后,接着用无水硫酸钠或无水硫酸镁干燥,最后,旋转蒸发干有机相得褐色固体,此时褐色固体为sonogashira反应的粗产品,包含目标产物、自身耦合副产物以及其他副产物。搭建好提取装置,在索氏提取器的提取瓶内倒入石油醚;将上述褐色固体装于滤纸筒里面,将滤纸筒放置在索氏提取器的提取管;加热回流1-2小时,将提取瓶的石油醚冷却至室温或更低,产物固体析出,利用抽滤装置过滤,获得纯度95%以上产物。
索氏提取法与柱层析色谱法对比:
本发明提供的苯乙炔类液晶材料的制备方法,操作方便,仪器装置简单,只需索氏提取装置和使用抽滤装置即可;节省溶剂,利用索氏提取器,相比柱层析色谱法只需较少的溶剂即可完成抽提;提纯损失少,产率在一般70%以上,比柱层析色谱法的产率高;获取的产物纯度高,普遍在95%以上;由于仪器,步骤简单,所需溶剂少,耗费时间少等优势,因此,此方法在工业化的大量生产中具有可行性。
实施例8
本实施例为的制备:
步骤1),制备最终产物:
搭建回流反应装置,向反应瓶内放入搅拌子,加入4-戊氧基环己基乙炔、1,2-二氟-4-溴苯、催化剂Pd(Pph3)2Cl2、碘化亚铜;将密封系统里的空气置换成氩气;注入三乙胺、四氢呋喃,搅拌并慢慢加热回流,确保系统在氩气保护下反应。5小时后,用TLC检测反应程度。
步骤2),用索式提取器对步骤1)粗产品进行提纯:
反应完毕后,将反应液倒入冰水,滴加稀盐酸并搅拌,滴至弱碱性;用二氯甲烷萃取上述液体,收集有机相,所得有机相用盐水洗两次后,接着用无水硫酸钠或无水硫酸镁干燥,最后,旋转蒸发干有机相得褐色固体,此时褐色固体为sonogashira反应的粗产品,包含目标产物、自身耦合副产物以及其他副产物。搭建好提取装置,在索氏提取器的提取瓶内倒入石油醚;将上述褐色固体装于滤纸筒里面,将滤纸筒放置在索氏提取器的提取管;加热回流1-2小时,将提取瓶的石油醚冷却至室温或更低,产物固体析出,利用抽滤装置过滤,获得纯度95%以上产物。
索氏提取法与柱层析色谱法对比:
上述仅为本发明的优选实施例而已,并不用于限制本发明,对于本领域的技术人员来说,本发明可以有各种更改和变化。凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!