一种溶剂黄124的合成方法与流程
本发明涉及溶剂染料合成领域,具体涉及一种溶剂黄124的合成方法。
背景技术:
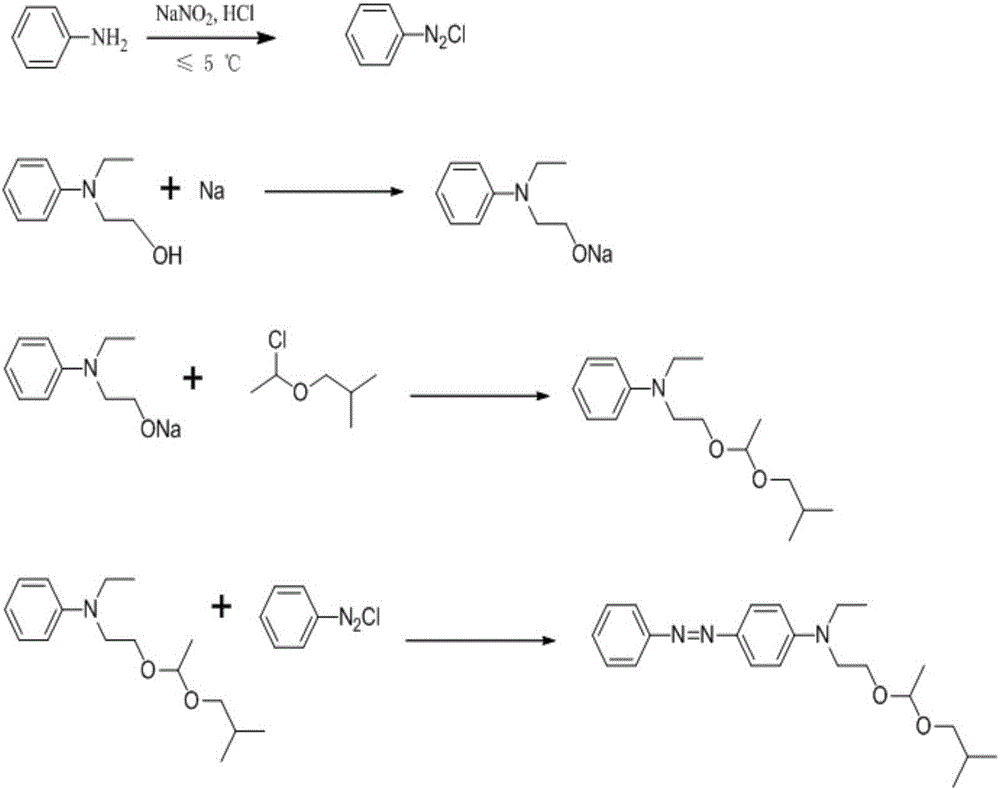
:染料是指在一定介质中,能使纤维或其他物质牢固着色的化合物。目前,染料已不只限于纺织物的染色和印花,在它在油漆、塑料、纸张、皮革、光电通讯、食品等许多部门得以应用。而溶剂染料是以能溶于有机溶剂为特征的一类染料。在这类染料结构中不含或少含亲水性基团。为了增大在溶剂中的溶解度常在染料结构中引入长碳链的烷基物,使染料具有电中性,在非纤维着色方面应用广泛。溶剂黄124,是一个偶氮结构的染料,其中含有不对称的缩醛结构,是一种性能优良的溶剂染料,主要用于塑料制品的作色,亦可经处理后作为分散染料用于涤纶及其混纺织物的染色。由于工艺条件的限制,目前其市售产品主含量偏低,质量不稳定,同时由于生产工艺收率较低造成单价较高,使其应用受到一定限制。专利号为US4011209的美国专利,公开了一种溶剂黄124的合成路线,以苯胺为原料,重氮化得到氯化重氮基苯后,经耦合反应后,再在三氯化硼的催化下进行缩醛反应制备得到溶剂黄124,该反应路线合成收率较低,副产物较多,同时收率较低。技术实现要素:为了解决上述的技术问题,本发明通过采用苯胺为原料经重氮化后得到氯化重氮基苯,将N-乙基-N-羟乙基苯胺和经处理后的金属钠颗粒反应得到醇钠,该醇钠经缩醛反应后,再和氯化重氮基苯进行耦合反应,制备得溶剂黄124,该反应路线合成收率高,制备出的产物纯度高。本发明采用如下技术方案:一种溶剂黄124的合成方法,包括以下步骤:1)氯化重氮基苯的制备在反应容器中加入苯胺、蒸馏水和浓硫酸,加热溶解,迅速冷却至5℃,缓慢滴加浓度为30%的亚硝酸钠溶液,在温度≤10℃,pH≤2下反应40-60min,TLC检测反应终点;反应结束后,加入尿素分解过量的亚硝酸钠,得绛红色重氮盐溶液,用浓盐酸调节该溶液pH至2.0-3.0,待用;2)N-乙基-N-羟乙基苯胺钠盐的制备将金属钠表面的煤油擦干后,切割成直径为0.5-1cm的颗粒,置入装有N-乙基-N-羟乙基苯胺的四氢呋喃溶液中,加热回流2-6h,降至室温,待用;3)N-乙基-N-(2-(1-异丁氧基乙氧基)乙基)苯胺的制备往装有步骤2)待用的N-乙基-N-羟乙基苯胺钠盐的反应容器中加入α-氯乙基异丁基醚,以四氢呋喃为溶液,加热回流4-5h,反应完毕降至室温,真空旋干四氢呋喃,再加入20%的氯化钠溶液,用乙酸乙酯萃取,收集乙酸乙酯层,回收至干得到油状物,加入丙酮溶液,待用;4)溶剂黄124的合成将步骤3)待用的N-乙基-N-(2-(1-异丁氧基乙氧基)乙基)苯胺的丙酮溶液冷却至3℃,加入有机碱调至pH=7.0~9.5,在5~10℃的条件下,在30min内滴入步骤1)制得的氯化重氮基苯溶液,搅拌2h。盐酸溶液调至pH=2.0-5.0,60℃加热15min后冷却至室温。加入乙酸乙酯萃取,收集的乙酸乙酯层,经饱和食盐水洗涤,无水硫酸镁干燥后、过滤,真空旋干溶剂,得到溶剂黄124。优选的,所述步骤1)中反应条件为在温度≤5℃,pH≤1下进行。进一步的,所述步骤1)中加入的亚硝酸钠总量与苯胺比例,按照摩尔比,亚硝酸钠:苯胺=1.2-1.4:1。优选的,所述步骤2)中反应时间为4-5h。优选的,所述步骤3)中真空旋干四氢呋喃的条件为:在真空度为0.07-0.09MPa,温度为40-50℃下回收。进一步的,所述步骤2)-步骤3)中,所使用的金属钠颗粒、N-乙基-N-羟乙基苯胺、α-氯乙基异丁基醚,按照摩尔比,金属钠颗粒:N-乙基-N-羟乙基苯胺:α-氯乙基异丁基醚=1:1.2-1.3:1.1-1.2。优选的,所述步骤4)中,所述的有机碱为三乙胺、三正丁胺、DBU中的任一种。优选的,所述步骤4)中,有机碱调至pH=7.5-8.5。优选的,所述步骤4)中,盐酸溶液调至pH=4.0-4.5。与现有技术比较,本发明具有以下优点:1、重氮化反应稳定,而且收率高,得到的氯化重氮基苯纯度高,为耦合反应提供了较好的基础。2、现有技术是先进行耦合反应,然后再进行缩醛反应,该缩醛反应条件是在酸性下进行的,会使得溶剂黄124的缩醛结构发生分解,易生成杂质,且该杂质不易分离;而本发明是先进行的缩醛反应,再在碱性条件下进行耦合反应,避免了溶剂黄124在酸性条件下发生分解的过程,同时本发明的选择性好,生成的杂质较少,从而收率高,产品纯度高。3、本发明合成过程中,反应条件温和,得到的中间体纯度较好,(略经后处理)可以直接用于进行下一步反应,为工业化的连续生产提供了技术支持。经本法合成的溶剂黄124的总收率达到68%,大大高于文献报道方法合成的总收率。有较高的应用价值,可以大幅度降低生产成本,提高产率和经济效益。附图说明图1为本发明合成方法涉及的反应方程式图2为国外标样溶剂黄124的HPLC图谱(450nm和320nm的条件下)图3为本发明实施例1所制备出的溶剂黄124的HPLC图谱(450nm和320nm的条件下)图4为国外标样溶剂黄124的紫外-可见吸收光谱图5为本发明实施例1所制备出的溶剂黄124的的紫外-可见吸收光谱具体实施方式以下结合实施例及附图对本发明的技术方案作进一步描述,但要求保护的范围并不局限于所述。实施例1一种溶剂黄124的合成方法,包括以下步骤:1)氯化重氮基苯的制备在反应容器中加入10g苯胺(0.1mol)、50ml蒸馏水和10ml浓硫酸,加热溶解,迅速冷却至5℃,缓慢滴加浓度为30%的亚硝酸钠溶液(总重为27.5g),在温度≤10℃,pH≤2下反应40min,TLC检测反应终点;反应结束后,加入2g尿素分解过量的亚硝酸钠,得绛红色重氮盐溶液,用浓盐酸调节该溶液pH至2.0-3.0,待用;2)N-乙基-N-羟乙基苯胺钠盐的制备将金属钠表面的煤油擦干后,切割成直径为0.5-1cm的颗粒,将28.43g的N-乙基-N-羟乙基苯胺(0.17mol)和切割好的3.25g金属钠颗粒(0.14mol)及200ml四氢呋喃加入反应容器后,加热回流2h,降至室温,待用;3)N-乙基-N-(2-(1-异丁氧基乙氧基)乙基)苯胺的制备往装有步骤2)待用的N-乙基-N-羟乙基苯胺钠盐的反应容器中加入21.50g的α-氯乙基异丁基醚(0.15mol),加入100ml四氢呋喃,加热回流4.5h,反应完毕降至室温,在真空度为0.07-0.09MPa,温度为40-50℃下旋干四氢呋喃,再加入100ml的20%的氯化钠溶液,用乙酸乙酯溶液萃取4次,每次用量为50ml,收集乙酸乙酯层,回收至干得到油状物,加入200ml的丙酮溶液,待用。4)溶剂黄124的合成将步骤3)待用的N-乙基-N-(2-(1-异丁氧基乙氧基)乙基)苯胺的丙酮溶液冷却至3℃,加入三乙胺调至pH=7.0~7.5,在5~10℃的条件下,在30min内滴入步骤1)制得的氯化重氮基苯溶液,搅拌2h。盐酸溶液调至pH=2.0-3.0,60℃加热15min后冷却至室温。加入乙酸乙酯萃取,再经饱和食盐水洗涤,30g无水硫酸镁干燥、过滤,真空旋干溶剂,得到溶剂黄124。实施例2一种溶剂黄124的合成方法,包括以下步骤:1)氯化重氮基苯的制备在反应容器中加入10g苯胺(0.1mol)、50ml蒸馏水和10ml浓硫酸,加热溶解,迅速冷却至5℃,缓慢滴加浓度为30%的亚硝酸钠溶液(总重为32.2g),在温度≤5℃,pH≤1下反应50min,TLC检测反应终点;反应结束后,加入2g尿素分解过量的亚硝酸钠,得绛红色重氮盐溶液,用浓盐酸调节该溶液pH至2.0-3.0,待用;2)N-乙基-N-羟乙基苯胺钠盐的制备将金属钠表面的煤油擦干后,切割成直径为0.5-1cm的颗粒,将30g的N-乙基-N-羟乙基苯胺(0.18mol)和切割好的3.25g金属钠颗粒(0.14mol)及200ml四氢呋喃加入反应容器后,加热回流4h,降至室温,待用;3)N-乙基-N-(2-(1-异丁氧基乙氧基)乙基)苯胺的制备往装有步骤2)待用的N-乙基-N-羟乙基苯胺钠盐的反应容器中加入22.50g的α-氯乙基异丁基醚(0.16mol),加入100ml四氢呋喃,加热回流4h,反应完毕降至室温,在真空度为0.07-0.09MPa,温度为40-50℃下旋干四氢呋喃,再加入100ml的20%的氯化钠溶液,用乙酸乙酯溶液萃取4次,每次用量为50ml,收集乙酸乙酯层,回收至干得到油状物,加入200ml的丙酮溶液,待用。4)溶剂黄124的合成将步骤3)待用的N-乙基-N-(2-(1-异丁氧基乙氧基)乙基)苯胺的丙酮溶液冷却至3℃,加入三乙胺调至pH=7.5~8.5,在5~10℃的条件下,在30min内滴入步骤1)制得的氯化重氮基苯溶液,搅拌2h。盐酸溶液调至pH=3.5-4.5,60℃加热15min后冷却至室温。加入乙酸乙酯萃取,再经饱和食盐水洗涤,30g无水硫酸镁干燥、过滤,真空旋干溶剂,得到溶剂黄124。实施例3一种溶剂黄124的合成方法,包括以下步骤:1)氯化重氮基苯的制备在反应容器中加入10g苯胺(0.1mol)、50ml蒸馏水和10ml浓硫酸,加热溶解,迅速冷却至5℃,缓慢滴加浓度为30%的亚硝酸钠溶液(总重为30g),在温度≤10℃,pH≤1下反应60min,TLC检测反应终点;反应结束后,加入2g尿素分解过量的亚硝酸钠,得绛红色重氮盐溶液,用浓盐酸调节该溶液pH至2.0-3.0,待用;2)N-乙基-N-羟乙基苯胺钠盐的制备将金属钠表面的煤油擦干后,切割成直径为0.5-1cm的颗粒,将31.30g的N-乙基-N-羟乙基苯胺(0.19mol)和切割好的3.25g金属钠颗粒(0.14mol)及200ml四氢呋喃加入反应容器后,加热回流6h,降至室温,待用;3)N-乙基-N-(2-(1-异丁氧基乙氧基)乙基)苯胺的制备往装有步骤2)待用的N-乙基-N-羟乙基苯胺钠盐的反应容器中加入22.50g的α-氯乙基异丁基醚(0.17mol),加入100ml四氢呋喃,,加热回流4.5h,反应完毕降至室温,在真空度为0.07-0.09MPa,温度为40-50℃下旋干四氢呋喃,再加入100ml的20%的氯化钠溶液,用乙酸乙酯溶液萃取4次,每次用量为50ml,收集乙酸乙酯层,回收至干得到油状物,加入200ml的丙酮溶液,待用。4)溶剂黄124的合成将步骤3)待用的N-乙基-N-(2-(1-异丁氧基乙氧基)乙基)苯胺的丙酮溶液冷却至3℃,加入三乙胺调至pH=8.5~9.5,在5~10℃的条件下,在30min内滴入步骤1)制得的氯化重氮基苯溶液,搅拌2h。盐酸溶液调至pH=4.5-5.0,60℃加热15min后冷却至室温。加入乙酸乙酯萃取,再经饱和食盐水洗涤,30g无水硫酸镁干燥、过滤,真空旋干溶剂,得到溶剂黄124。实施例4一种溶剂黄124的合成方法,包括以下步骤:1)氯化重氮基苯的制备在反应容器中加入10g苯胺(0.1mol)、50ml蒸馏水和10ml浓硫酸,加热溶解,迅速冷却至5℃,缓慢滴加浓度为30%的亚硝酸钠溶液(总重为27.5g),在温度≤5℃,pH≤2下反应60min,TLC检测反应终点;反应结束后,加入2g尿素分解过量的亚硝酸钠,得绛红色重氮盐溶液,用浓盐酸调节该溶液pH至2.0-3.0,待用;2)N-乙基-N-羟乙基苯胺钠盐的制备将金属钠表面的煤油擦干后,切割成直径为0.5-1cm的颗粒,将28.43g的N-乙基-N-羟乙基苯胺(0.17mol)和切割好的3.25g金属钠颗粒(0.14mol)及200ml四氢呋喃加入反应容器后,加热回流5h,降至室温,待用;3)N-乙基-N-(2-(1-异丁氧基乙氧基)乙基)苯胺的制备往装有步骤2)待用的N-乙基-N-羟乙基苯胺钠盐的反应容器中加入22.50g的α-氯乙基异丁基醚(0.16mol),加入100ml四氢呋喃,加热回流4h,反应完毕降至室温,在真空度为0.07-0.09MPa,温度为40-50℃下旋干四氢呋喃,再加入100ml的20%的氯化钠溶液,用乙酸乙酯溶液萃取4次,每次用量为50ml,收集乙酸乙酯层,回收至干得到油状物,加入200ml的丙酮溶液,待用。4)溶剂黄124的合成将步骤3)待用的N-乙基-N-(2-(1-异丁氧基乙氧基)乙基)苯胺的丙酮溶液冷却至3℃,加入三正丁胺调至pH=8.5~9.5,在5~10℃的条件下,在30min内滴入步骤1)制得的氯化重氮基苯溶液,搅拌2h。盐酸溶液调至pH=4.0-4.5,60℃加热15min后冷却至室温。加入乙酸乙酯萃取,再经饱和食盐水洗涤,30g无水硫酸镁干燥、过滤,真空旋干溶剂,得到溶剂黄124。实施例5一种溶剂黄124的合成方法,包括以下步骤:1)氯化重氮基苯的制备在反应容器中加入10g苯胺(0.1mol)、50ml蒸馏水和10ml浓硫酸,加热溶解,迅速冷却至5℃,缓慢滴加浓度为30%的亚硝酸钠溶液(总重为30g),在温度≤5℃,pH≤1下反应50min,TLC检测反应终点;反应结束后,2g尿素分解过量的亚硝酸钠,得绛红色重氮盐溶液,用浓盐酸调节该溶液pH至2.0-3.0,待用;2)N-乙基-N-羟乙基苯胺钠盐的制备将金属钠表面的煤油擦干后,切割成直径为0.5-1cm的颗粒,将31.30gg的N-乙基-N-羟乙基苯胺(0.19mol)和切割好的3.25g金属钠颗粒(0.14mol)及200ml四氢呋喃加入反应容器后,加热回流4.5h,降至室温,待用;3)N-乙基-N-(2-(1-异丁氧基乙氧基)乙基)苯胺的制备往装有步骤2)待用的N-乙基-N-羟乙基苯胺钠盐的反应容器中加入23.50g的α-氯乙基异丁基醚(0.17mol),加入100ml四氢呋喃,加热回流4.5h,反应完毕降至室温,在真空度为0.07-0.09MPa,温度为40-50℃下旋干四氢呋喃,再加入100ml的20%的氯化钠溶液,用乙酸乙酯溶液萃取4次,每次用量为50ml,收集乙酸乙酯层,回收至干得到油状物,加入200ml的丙酮溶液,待用。4)溶剂黄124的合成将步骤3)待用的N-乙基-N-(2-(1-异丁氧基乙氧基)乙基)苯胺的丙酮溶液冷却至3℃,加入三正丁胺调至pH=7.5~8.5,在5~10℃的条件下,在30min内滴入步骤1)制得的氯化重氮基苯溶液,搅拌2h。盐酸溶液调至pH=2.0-3.0,60℃加热15min后冷却至室温。加入乙酸乙酯萃取,再经饱和食盐水洗涤,30g无水硫酸镁干燥、过滤,真空旋干溶剂,得到溶剂黄124。实施例6一种溶剂黄124的合成方法,包括以下步骤:1)氯化重氮基苯的制备在反应容器中加入10g苯胺(0.1mol)、50ml蒸馏水和10ml浓硫酸,加热溶解,迅速冷却至5℃,缓慢滴加浓度为30%的亚硝酸钠溶液(总重为33.0g),在温度≤10℃,pH≤1下反应40min,TLC检测反应终点;反应结束后,加入2g尿素分解过量的亚硝酸钠,得绛红色重氮盐溶液,用浓盐酸调节该溶液pH至2.0-3.0,待用;2)N-乙基-N-羟乙基苯胺钠盐的制备将金属钠表面的煤油擦干后,切割成直径为0.5-1cm的颗粒,将30g的N-乙基-N-羟乙基苯胺(0.18mol)和切割好的3.25g金属钠颗粒(0.14mol)及200ml四氢呋喃加入反应容器后,加热回流6h,降至室温,待用;3)N-乙基-N-(2-(1-异丁氧基乙氧基)乙基)苯胺的制备往装有步骤2)待用的N-乙基-N-羟乙基苯胺钠盐的反应容器中加入21.50g的α-氯乙基异丁基醚(0.15mol),加入100ml四氢呋喃,加热回流4h,反应完毕降至室温,在真空度为0.07-0.09MPa,温度为40-50℃下旋干四氢呋喃,再加入100ml的20%的氯化钠溶液,用乙酸乙酯溶液萃取4次,每次用量为50ml,收集乙酸乙酯层,回收至干得到油状物,加入200ml的丙酮溶液,待用。4)溶剂黄124的合成将步骤3)待用的N-乙基-N-(2-(1-异丁氧基乙氧基)乙基)苯胺的丙酮溶液冷却至3℃,加入DBU调至pH=7.0~8.0,在5~10℃的条件下,在30min内滴入步骤1)制得的氯化重氮基苯溶液,搅拌2h。盐酸溶液调至pH=4.5-5.0,60℃加热15min后冷却至室温。加入乙酸乙酯萃取,再经饱和食盐水洗涤,30g无水硫酸镁干燥、过滤,真空旋干溶剂,得到溶剂黄124。实施例7一种溶剂黄124的合成方法,包括以下步骤:1)氯化重氮基苯的制备在反应容器中加入10g苯胺(0.1mol)、50ml蒸馏水和10ml浓硫酸,加热溶解,迅速冷却至5℃,缓慢滴加浓度为30%的亚硝酸钠溶液(总重为28g),在温度≤10℃,pH≤2下反应60min,TLC检测反应终点;反应结束后,加入2g尿素分解过量的亚硝酸钠,得绛红色重氮盐溶液,用浓盐酸调节该溶液pH至2.0-3.0,待用;2)N-乙基-N-羟乙基苯胺钠盐的制备将金属钠表面的煤油擦干后,切割成直径为0.5-1cm的颗粒,将28.50g的N-乙基-N-羟乙基苯胺(0.17mol)和切割好的3.25g金属钠颗粒(0.14mol)及200ml四氢呋喃加入反应容器后,加热回流2h,降至室温,待用;3)N-乙基-N-(2-(1-异丁氧基乙氧基)乙基)苯胺的制备往装有步骤2)待用的N-乙基-N-羟乙基苯胺钠盐的反应容器中加入21.50的α-氯乙基异丁基醚(0.15mol),加入100ml四氢呋喃,加热回流5h,反应完毕降至室温,在真空度为0.07-0.09MPa,温度为40-50℃下旋干四氢呋喃,再加入100ml的20%的氯化钠溶液,用乙酸乙酯溶液萃取4次,每次用量为50ml,收集乙酸乙酯层,回收至干得到油状物,加入200ml的丙酮溶液,待用。4)溶剂黄124的合成将步骤3)待用的N-乙基-N-(2-(1-异丁氧基乙氧基)乙基)苯胺的丙酮溶液冷却至3℃,加入DBU调至pH=8.5~9.5,在5~10℃的条件下,在30min内滴入步骤1)制得的氯化重氮基苯溶液,搅拌2h。盐酸溶液调至pH=2.0-3.0,60℃加热15min后冷却至室温。加入乙酸乙酯萃取,再经饱和食盐水洗涤,30g无水硫酸镁干燥、过滤,真空旋干溶剂,得到溶剂黄124。对实施例1-实施例7制备出的溶剂黄124的收率和质量情况统计分析,同时也按照US4011209的实施例,进行溶剂黄124的制备,结果如下表所示:项目总摩尔收率(%)色谱纯度(%)总杂(%)实施例161.5093.255.38实施例268.2496.792.05实施例359.3993.155.10实施例469.1797.011.68实施例568.4696.222.01实施例664.7894.303.89实施例762.6093.524.78US401120915.3085.1410.21备注:总摩尔收率,以苯胺为起始物料来进行计算。从上表的实验结果可以看出,本发明制备出的溶剂黄124,总摩尔收率高,而且色谱纯度高,总杂较低;同时在步骤1)的制备过程中发现,制备得到的氯化重氮基苯溶液,在冷藏条件下储存,1-2天内分解出的杂质较少,比较稳定,而相比专利US4011209制备出的氯化重氮基苯溶液,在相同的条件下储存,在4-6h后,较易分解,生产较多的杂质。同时对比本发明实施例和专利US4011209的实施例,发明本发明实施例所制备出的溶剂黄124,在塑料制品的作色方面,鲜艳度和色牢度远优于专利US4011209的实施例在在塑料制品的作色方面的效果。当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1