高效光催化复合材料及其制备方法与流程
本发明涉及一种光催化材料及其制备方法,尤其是涉及一种具有高比表面的增强剂辅助的高效光催化剂及其制备方法。
背景技术:
现有主要的光催化剂产品通常存在着效率低下、有效接触面积低下等问题。其主要原因在于:1.光催剂本身的比表面积有限,难以和待催化物进行充分接触;2.光催化剂的电子空穴分离效率较低;3.光照辐射的利用效率低下。
故如何开发出高效的光催化剂材料及其制备方法是目前亟需解决的问题。
技术实现要素:
本发明提出一种高效光催化复合材料及其制备方法,解决了现有技术中光催化材料效率低下的问题。这种复合材料复合水净化和空气净化的技术要求,且其制备工艺简单易于大规模生产。
本发明的技术方案是这样实现的:一种高效光催化复合材料,至少包含载体、光催化剂单体和增强剂颗粒,所述光催化剂单体是具备压电效应的半导体;所述增强剂颗粒是磁性纳米颗粒或/和具备局域表面等离子共振效应的金属纳米颗粒;光催化剂单体在载体上分布形成光催化剂阵列,增强剂颗粒分布在光催化剂阵列上形成增强剂阵列。
所述光催化剂单体的形貌为核壳结构、嵌段结构、异质结、半封闭结构、封闭结构、实心结构或其组合。
一种高效光催化复合材料的制备方法,是按照下述步骤进行的:
(1)将载体进行表面活化处理,而后将载体表面喷涂上一层尺寸为1~100nm的ZnO纳米晶种,并在50~100℃温度下进行干燥0.5~12小时;
将硝酸锌、六次甲基四胺及氨水加入去离子水中搅拌均匀,混合成为前驱体溶液;
将干燥后的负载有ZnO晶种的载体搁置进入该前驱体溶液中,在60~90℃的温度下,静置反应5~15小时,在载体表面形成氧化锌纳米阵列,得到载体—光催化剂阵列结构;
(2)将5~100nm磁性纳米颗粒、柠檬酸三钠加入去离子水中,其中磁性纳米颗粒、柠檬酸三钠及水的质量比为1:4~16:40~150,将混合溶液在50~80℃搅拌反应3~24小时;反应完毕后,将反应后的磁性纳米颗粒产物用磁铁收集,并用去离子水清洗2~3次,将磁性纳米颗粒产物分散在去离子水中,由此得到浓度为0.0001~0.01g/ml的柠檬酸盐修饰的磁性纳米颗粒分散液;
将步骤(1)所获得的载体—光催化剂阵列结构,浸泡进入磁性纳米颗粒分散液中,浸泡24小时后取出;将浸泡完的产物,在200~450℃下,惰性气体保护处理0.5~10小时,得到载体—光催化剂阵列—磁性纳米颗粒阵列。
将步骤(2)获得的产物,浸泡进入摩尔浓度为0.01~1M的金属盐溶液中,并用紫外灯辐照,金属盐在光催化剂阵列表面还原形成金属单质颗粒阵列,由此构成载体—光催化剂阵列—磁性纳米颗粒-金属颗粒阵列。
所述ZnO纳米颗粒的尺寸为20nm,干燥温度为70℃,干燥时间为2小时。
所述载体为石墨烯、玻璃纤维、陶粒、石英纤维、光纤、气凝胶、泡沫金属、薄膜或其组合。
所述磁性纳米颗粒为包含Fe、Co、Ni、Cr元素中的合金、氧化物或单质。
所述金属盐中的金属元素为Au、Ag、Cu、Fe、Sn、Pt或Pd中的至少一种。
本发明中,金属颗粒通过表面等离子体共振效应提高光催化效率;磁性颗粒,磁性颗粒在外加磁场下受磁力作用,并将磁力传递于具备压电效应的半导体阵列使其发生形变,由此改变半导体的能级结构,提高光催化效率。本发明的优点是,(1)较大的光催化接触面积;(2)较高的光催化效率;(3)简易的制备方法,易于大规模实施。
附图说明
为了更清楚地说明本发明实施例或现有技术中的技术方案,下面将对实施例或现有技术描述中所需要使用的附图作简单地介绍,显而易见地,下面描述中的附图仅仅是本发明的一些实施例,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据这些附图获得其他的附图。
图1(a)、1(b)、1(c)和1(d)非限制性光催化剂阵列在载体上的分布示意图。
图2(a)和2(b)为非限制性光线在光催化剂表面的多次折射模型示意图。
图3(a)、3(b)、3(c)、3(d)、3(e)、3(f)、3(g)和3(h)为非限制性光催化剂形貌结构示意图。
图4(a)、4(b)、4(c)为非限制性增强剂颗粒阵列在光催化剂阵列上的分布示意图。
图5为非限制性磁力驱动的光催化剂形变示意图。
图6为非限制性增强剂以组合方式提高光催化效率的示意图。
图7为纤维载体—致密排列锥形氧化锌阵列结构的扫描电子显微镜图片
图8(a)和(b)为纤维载体—疏松排列锥形氧化锌阵列结构的扫描电子显微镜图片
图9(a)和(b)为纤维载体—聚集型致密排列锥形氧化锌阵列结构的扫描电子显微镜图片。
图10为纤维载体—球形二氧化钛结构的扫描电子显微镜图片。
图11为纤维载体—聚集型致密排列氧化锌阵列—金纳米颗粒阵列结构的扫描电子显微镜图片。
图12为纤维载体—聚集型致密排列氧化锌阵列—银纳米颗粒阵列结构的扫描电子显微镜图片。
图13为纤维载体—聚集型致密排列氧化锌阵列—四氧化三铁纳米颗粒阵列结构的扫描电子显微镜图片。
具体实施方式
下面将结合本发明实施例中的附图,对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有付出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
一种高效光催化复合材料,至少包含载体、光催化剂单体和增强剂颗粒,所述光催化剂单体是具备压电效应的半导体;所述增强剂颗粒是磁性纳米颗粒或/和具备局域表面等离子共振效应的金属纳米颗粒;光催化剂单体在载体上分布形成光催化剂阵列,增强剂颗粒分布在光催化剂阵列上形成增强剂阵列。
所述光催化剂单体其形貌包括:核壳结构、嵌段结构、异质结、半封闭结构、封闭结构、实心结构或其组合。
一种高效光催化复合材料的制备方法,是按照下述步骤进行的:
(1)将载体进行表面活化处理,而后将载体表面喷涂上一层尺寸为1~100nm的ZnO纳米晶种,并在50~100℃温度下进行干燥0.5~12小时;干燥温度可以为50、60、75、90或100℃,根据需要可以调整干燥时间为12、10、7、4、2.5或0.5h。
将硝酸锌、六次甲基四胺及氨水加入去离子水中搅拌均匀,混合成为前驱体溶液,其中硝酸锌浓度为5~20mM、六次甲基四胺浓度为5~20mM,氨水占混合溶液的体积比为1~20%;前驱体溶液中、六次甲基四胺的浓度可调,比如采用下述三种实施方案:1.硝酸锌浓度为5mM、六次甲基四胺浓度为5mM,氨水占混合溶液的体积比为1%;2.硝酸锌浓度为10mM、六次甲基四胺浓度为10mM,氨水占混合溶液的体积比为10%;3.硝酸锌浓度为20mM、六次甲基四胺浓度为20mM,氨水占混合溶液的体积比为20%,或者其他方式。
将干燥后的负载有ZnO晶种的载体搁置进入该前驱体溶液中,在60~90℃的温度下,静置反应5~15小时,在载体表面形成氧化锌纳米阵列,得到载体—光催化剂阵列结构;
(2)将5~100nm磁性纳米颗粒、柠檬酸三钠加入去离子水中,其中磁性纳米颗粒、柠檬酸三钠及水的质量比为1:4~16:40~150,将混合溶液在50~80℃搅拌反应3~24小时;反应完毕后,将反应后的磁性纳米颗粒产物用磁铁收集,并用去离子水清洗2~3次,将磁性纳米颗粒产物分散在去离子水中,由此得到浓度为0.0001~0.01g/ml的柠檬酸盐修饰的磁性纳米颗粒分散液;
将步骤(1)所获得的载体—光催化剂阵列结构,浸泡进入磁性纳米颗粒分散液中,浸泡24小时后取出;将浸泡完的产物,在200~450℃下,惰性气体保护处理0.5~10小时,得到载体—光催化剂阵列—磁性纳米颗粒阵列;
进一步的,将步骤(2)获得的产物,浸泡进入摩尔浓度为0.01~1M的金属盐溶液中,并用紫外灯辐照,金属盐在光催化剂阵列表面还原形成金属单质颗粒阵列,载体—光催化剂阵列—磁性纳米颗粒-金属颗粒阵列,由此构成高效光催化复合材料。
优选的,所述ZnO纳米颗粒的尺寸为20nm,干燥温度为70℃,干燥时间为2小时。
光催化剂通常和待催化物难以形成充分的有效接触,因此载体是重要的提高比表面积的材料。通常载体可以通过相互缠绕形成三维结构,例如玻璃纤维通过缠绕形成玻璃棉、石英纤维通过相互缠绕形成石英棉、石墨烯通过相互缠绕联接形成石墨烯气凝胶等。光催化剂在三维载体上形成空间结构,对于反应物的接触是有利的。本申请所述载体为石墨烯、玻璃纤维、陶粒、石英纤维、光纤、气凝胶、泡沫金属、薄膜或其组合。
所述磁性纳米颗粒为包含Fe、Co、Ni、Cr元素中的合金、氧化物或单质。
所述金属盐中的金属元素为Au、Ag、Cu、Fe、Sn、Pt或Pd中的至少一种。通常金属盐通过光催化作用在光催化剂表面原位还原得到金属纳米颗粒,其基本原理可以参考文献:Lu L,Wohlfart A,Parala H,et al.A novel preparation of nano-Cu/ZnO by photo-reduction of Cu(OCH(Me)CH2NMe2)2on ZnO at room temperature.[J].Chemical Communications,2003,1(1):40-1。
所述金属纳米颗粒阵列具备抗菌特性,这一点对于光催化剂在空气净化和水处理方向的应用是有利的。例如选择金属颗粒的化学构成为银元素,银通常具有较好的抗菌效果。
载体的活化处理通常用于增加附着力,例如通过N2Plasmon(等离子体振子)轰击处理载体表面;另外,载体的活化处理也可以用于增加比表面积,例如泡沫合金作为载体,用酸性腐蚀液腐蚀掉另一金属相。步骤(1)中的活化处理通常包括但不限于:高能粒子流轰击、高能射线轰击、激光刻蚀、化学腐蚀、Plasmon处理或其组合。在下述实施例中主要采用了N2plasmon轰击的方式进行载体表面活化。
通常光催化剂单体构成的阵列结构是规则的,如图1(a)展示了锥形光催化剂构成的规则阵列结构,光催化剂阵列101生长在载体201上。通常阵列中,光催化剂单体之间是可以相互接触的也可以是分离的,如图1(b)展示了锥形光催化剂单体相互接触的阵列,其中光催化剂阵列101单体之间有的是接触,有的是分离。另外,光催化剂阵列101在载体201上构筑的阵列可选的是随机分布的,如图1(c)所示。在更为普遍的结构中,光催化剂阵列各单体之间的形貌是不相同的,例如图1(d)展示了锥形和颗粒两种形貌的光催化剂单体构成的光催化剂阵列,其中是锥形单体104和颗粒单体105均生长在载体201上的光催化剂阵列。
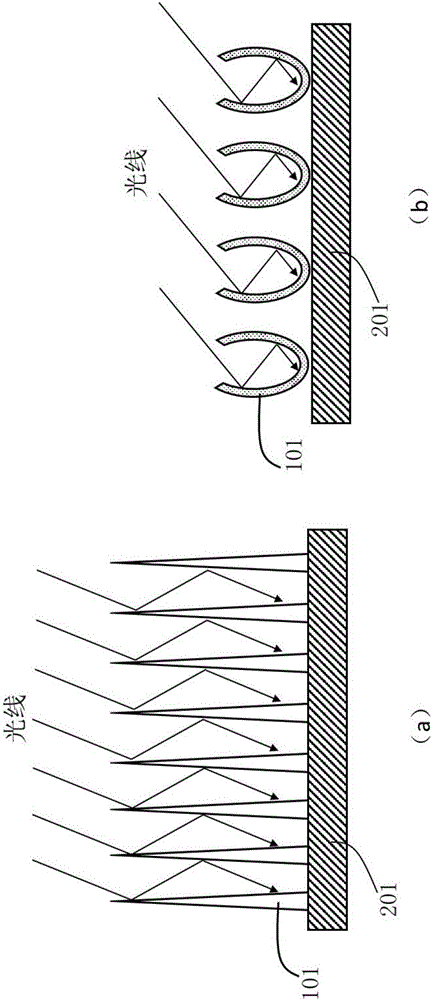
在本发明中,光催化剂阵列通常对于光线能够进行多次散射或者多次反射,这一特点确定了光催化剂对光线的高效吸收。如图2(a)展示了锥形光催化剂阵列单体之间相互反射所形成的光催化剂阵列;如图2(b)展示了半封闭光催化剂阵列单体内部进行多次光线反射提高光线吸收的示意图,光催化剂阵列101形成了半封闭结构。
通常光催化剂单体的形貌和结构是多样的,通常包括但不限于:核壳结构、嵌段结构、异质结、半封闭结构、封闭结构、实心结构或其组合。如图3(a)所示一种核壳结构的光催化剂单体,其中核108设置在109壳内,核壳具有不一致的化学构成,例如核是ZnO,壳是TiO2。如图3(b)展示了一种嵌段光催化剂结构,其中第一嵌段111的化学构成异于第二嵌段110和第三嵌段112的化学构成,通常对于光催化剂的电子空穴的分离效率具有提高的效应,类似于半导体结构中的PNP型或NPN型结构。如图3(c)展示了一种异质结结构的光催化剂单体,其中第一单体113和第一单体114的化学构成不同。半封闭结构的光催化剂单体示意图如图3(d)所示。全封闭结构的光催化剂单体示意图如3(e)所示,例如空心结构通常是一个比较普遍的全封闭结构。实体结构的光催化剂单体示意图如图3(f)所示,通常实体结构的光催化剂单体是最为常见的,例如本发明实施例中的展示的锥形ZnO单体结构。当然上述结构的任意组合也是本发明光催化剂单体所包括的形貌,例如3(g)展示了半封闭结构和异质结结构的结合;例如3(h)展示了封闭结构和实体结构的结合,实体颗粒116位于外壳115内,构成封闭结构。
光催化剂单体的形貌往往是依赖于光催化剂本身的晶体构成,例如ZnO光催化剂的晶体结构通常是六边纤锌矿结构,其形貌构成通常包括但不限于:锥形、棒状、塔状。
增强剂颗粒阵列是由增强剂单体按照随机分布或特定规律分布在光催化剂阵列之上构成。通常增强剂颗粒可以基于光催化剂单体本身构成阵列,也可以基于光催化剂阵列整体构成阵列。如图4(a)展示了增强剂颗粒阵列随机分布于的光催化剂阵列之上的非限制性示意图,其中包括载体201、光催化剂阵列101、第一增强剂粒单体301和第一增强剂粒单体302,第一增强剂粒单体301和第一增强剂粒单体302在光催化剂阵列上随机分布。如图4(b)展示了增强剂颗粒阵列在光催化剂单体上形成阵列的方式,其中增强剂颗粒单体303在光催化剂单体117形成阵列,金属颗粒单体304在光催化剂单体117上形成阵列,增强剂颗粒303构成的阵列与金属颗粒单体304构成的阵列可以具有同样的分布规律也可以具有不一致的分布规律。在一些实施方式中,增强剂颗粒单体同时和多个光催化剂单体接触形成阵列,一种非限制性示意方式如图4(c)所示,其中增强剂颗粒单体303与三个光催化剂单体117同时接触,由此构成增强剂颗粒阵列。
所述磁性纳米颗粒在外加磁场下受磁力作用,并将磁力传递于具备压电效应的半导体光催化剂阵列使其发生形变,由此改变半导体的能级结构。通常半导体光催化剂选择一维结构能够在外力作用下产生更大的形变,例如选择30nm的Fe3O4作为增强剂颗粒,选择锥形的ZnO纳米棒作为光催化剂阵列单体。Fe3O4纳米颗粒具有超顺磁性,对于外加磁场具有力学响应。ZnO是一类同时兼具压电效应、光催化效应的半导体,外力施加在其c轴方向时,产生压电效应半导体能级结构随之改变,相关机理可以参考文献:Wang Z L.Piezotronic and Piezophototronic Effects[J].Journal of Physical Chemistry Letters,2010,1(9):1388-1393.及Wang L,Liu S,Zheng W,et al.Piezotronic Effect Enhanced Photocatalysis in Strained Anisotropic ZnO/TiO2Nanoplatelets via Thermal Stress[J].Acs Nano,2016.10(2),pp 2636–2643。
一种非限制性实施方式如图5所示,在磁场的作用下Fe3O4纳米颗粒306受磁力作用牵引其所吸附的锥形ZnO纳米棒阵列120弯曲,锥形ZnO纳米棒阵列的能级结构由此改变,其对光子的吸收和转化效率随之改变,从而对光催化效率进行调节。
一种非限制性组合形式的增强结构如图6所示,在磁场的作用下Fe3O4纳米颗粒306受磁力作用牵引其所吸附的锥形ZnO纳米棒阵列120弯曲,锥形ZnO阵列的能级结构由此改变,另外具备局域表面等离子体共振效应的的银颗粒308在光照下能够有效的分离激发的空穴电子对,上述两种方式共同作用提高光催化效率。
在以下实施例中,用紫外/可见/近红外分光光度计(PerkinElmer Lambda950)测定光催化效率;测定复合材料的结构用扫描电子显微镜(SEM Hitach S4800)进行表征。
实施例1:纤维载体—致密排列锥形氧化锌阵列结构,制备方法如下:
(1)将纤维载体进行N2Plasmon处理;
(2)在纤维载体表面喷涂上一层ZnO纳米颗粒晶种,并在70℃温度下干燥12小时;
(3)将硝酸锌、六次甲基四胺及氨水混合得到前驱体溶液(其中硝酸锌浓度为10mM、六次甲基四胺为10mM,氨水浓度25%,氨水占混合溶液的体积比为4%),将步骤(2)获得的制品放入前驱体溶液于75℃左右静置反应12h,由此形成在载体表面形成锥形的氧化锌纳米阵列;得到载体—光催化剂阵列结构。其扫描电镜图如图7所示,可以看到,锥形氧化锌阵列在载体上形成致密排列。
本实施例中用到的纤维为玻璃纤维或者石英纤维。
实施例2:纤维载体—疏松排列锥形氧化锌阵列结构的制备方法:
(1)将纤维载体进行N2Plasmon处理;
(2)将纤维载体表面喷涂上一层ZnO纳米颗粒晶种,并在70℃温度下干燥12小时;
(3)将硝酸锌、六次甲基四胺及氨水混合得到前驱体溶液(其中硝酸锌浓度为10mM、六次甲基四胺为10mM,氨水占混合溶液的体积比为2%),将步骤(2)获得的制品放入前驱体溶液于75℃左右静置反应12h,由此形成锥形的氧化锌纳米阵列;得到载体—光催化剂阵列结构。其扫描电镜图如图8(a)(b)所示,可以看到锥形氧化锌阵列在载体上形成疏松排列。
本实施例中用到的纤维为玻璃纤维或者石英纤维。
实施例3:纤维载体—聚集型致密排列锥形氧化锌阵列结构制备方法如下:将实施例1中的所得制品,浸泡在0.5mg/ml的氧化石墨烯溶液1min,取出后将其自然晾干。其扫面电镜图如图9(a)和(b)所示,可以看到,由于石墨烯的存在,致密排列锥形氧化锌阵列的多个单体之间的尖端聚集在一起,形成纤维载体—聚集型致密排列锥形氧化锌阵列结构。
实施例4:纤维载体—球形二氧化钛结构,制备方法:
(1)将纤维载体进行O2Plasmon处理;
(2)将纤维载体表面喷涂钛酸四丁酯前驱体溶液;
(3)将步骤(2)所得制品放入马弗炉中,设置温度600℃煅烧5小时。
结构:其形貌结构如图10所示。
实施例5:纤维载体—聚集型致密排列氧化锌阵列—银纳米颗粒阵列结构
制备方法:(1)将纤维载体进行N2Plasmon处理;
(2)将纤维载体表面喷涂上一层ZnO纳米颗粒晶种,并在70℃的温度下进行干燥;
(3)将硝酸锌、六次甲基四胺及氨水混合得前驱体溶液(其中硝酸锌浓度为10mM、六次甲基四胺为10mM,氨水所占混合溶液的体积比为4%),将步骤(2)获得的制品放入前驱体容易,于75℃左右搁置12h,由此形成锥形的氧化锌纳米阵列由此,得到光催化剂阵列;
(4)将步骤(3)所获得制品,浸泡在1mM的AgNO3溶液中,并用紫外氙灯照射5min,AgNO3被氧化锌光催化还原成AgO;
(5)将步骤(4)所获得制品在300℃下,惰性气体保护,退火2小时,由此得到载体—光催化剂阵列—金属纳米颗粒阵列结构。其中步骤(1)~(3)与实施例1一致。
结构:其形貌结构如图11所示,银纳米颗粒在锥形氧化锌纳米阵列上形成次级结构阵列。
实施例6:纤维载体—聚集型致密排列氧化锌阵列—四氧化三铁纳米颗粒阵列结构
制备方法:(1)将纤维载体进行N2Plasmon处理;
(2)将纤维载体表面喷涂上一层ZnO纳米颗粒中子,并在70℃温度下干燥12小时;
(3)将(2)步骤获得的制品放入75℃下的硝酸锌、六次甲基四胺及氨水的混合前驱体溶液(其中硝酸锌浓度为10mM、六次甲基四胺为10mM,氨水占混合溶液的体积比为2%),搁置12h,由此锥形的氧化锌纳米阵列由此形成;得到载体—光催化剂阵列结构;
(4)将平均直径40nm的磁性纳米颗粒、柠檬酸三钠加入去离子水中,其中40nm磁性Fe3O4纳米颗粒、柠檬酸三钠及水的质量比为1:4~16:40~150,将混合溶液在80℃搅拌反应10小时;反应完毕后,将反应后的磁性纳米颗粒产物用磁铁收集,并用去离子水清洗2~3次,并将其分散在去离子水中,浓度为0.001g/ml,由此得到柠檬酸盐修饰的磁性纳米颗粒分散液;
(5)将(3)步骤所获得的载体—光催化剂阵列结构,浸泡进入(4)步骤柠檬酸盐修饰的磁性纳米颗粒分散液中,浸泡24小时后取出;将浸泡完的产物,在300℃下,惰性气体保护处理0.5~10小时,由此得到载体—光催化剂阵列—磁性纳米颗粒阵列结构;其中步骤(1)~(3)与实施例1一致。
结构:其形貌结构如图12所示,Fe3O4纳米颗粒在锥形氧化锌纳米阵列上形成,单个Fe3O4纳米颗粒和多个锥形氧化锌单体接触,使得氧化锌阵列变成聚集型氧化锌阵列。
实施例7:纤维载体—聚集型致密排列氧化锌阵列—四氧化三铁纳米颗粒阵列—银纳米颗粒阵列结构
制备方法:(1)将纤维载体进行N2Plasmon处理;
(2)将纤维载体表面喷涂上一层ZnO纳米颗粒中子,并在70℃温度下干燥12小时;
(3)将(2)步骤获得的制品放入75℃下的硝酸锌、六次甲基四胺及氨水的混合前驱体溶液(其中硝酸锌浓度为10mM、六次甲基四胺为10mM,氨水占混合溶液的体积比为2%),搁置12h,由此锥形的氧化锌纳米阵列由此形成;得到载体—光催化剂阵列结构;
(4)将平均直径40nm的磁性纳米颗粒、柠檬酸三钠加入去离子水中,其中40nm磁性Fe3O4纳米颗粒、柠檬酸三钠及水的质量比为1:4~16:40~150,将混合溶液在80℃搅拌反应10小时;反应完毕后,将反应后的磁性纳米颗粒产物用磁铁收集,并用去离子水清洗2~3次,并将其分散在去离子水中,浓度为0.001g/ml,由此得到柠檬酸盐修饰的磁性纳米颗粒分散液;
(5)将(3)步骤所获得的载体—光催化剂阵列结构,浸泡进入(4)步骤柠檬酸盐修饰的磁性纳米颗粒分散液中,浸泡24小时后取出;将浸泡完的产物,在300℃下,惰性气体保护处理50小时,由此得到载体—光催化剂阵列—磁性纳米颗粒阵列结构;
(6)将步骤(5)所获得制品,浸泡在1mM的AgNO3溶液中,并用紫外氙灯照射5min,AgNO3被氧化锌光催化还原成Ag0;
(7)将步骤(6)所获得制品在300摄氏度下,惰性气体保护,退火2小时,由此得到载体—光催化剂阵列—金属纳米颗粒阵列结构。其中步骤(1)~(3)与实施例1一致。
结构:其形貌结构如图13所示,增强剂颗粒在致密排列锥形氧化锌纳米阵列上形成,多个锥形氧化锌单体之间的尖端聚集在一起。
对比例1光催化效率
选取实施例1~7中所制备的光催化复合材料,并保证具有一致的重量,将上述光催化剂放入10PPM的甲基橙溶液中,加并在实施例1~7的光催化剂上辐照同样强度和同样波长的紫外光线,辐照时间均为半小时。通过紫外可见光谱测定甲基橙的催化前和催化后的归一化的吸收强度,判定其光催化效率高低,如下表所示
上述对比例证明增强剂颗粒对光催化效率具有提高作用。
以上所述仅为本发明的较佳实施例而已,并不用以限制本发明,凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!