氟氰戊菊酯分子印迹固相萃取柱的制备方法及在烟叶分析中的应用与流程
本发明专利涉及化工有机分析技术领域,具体是氟氰戊菊酯分子印迹固相萃取柱的制备方法及在烟叶分析中的应用。
背景技术:
氟氰戊菊酯是一种广谱性拟除虫菊酯类杀虫剂,近年来在我国已被广泛应用于预防烟草害虫。
按中国农药毒性分级标准,氟氰戊菊酯及其结构类似物属于低毒杀菌剂,雄性大鼠急性经口 LD50为81mg/kg,雌性为67mg/kg。超标使用必然会导致烟草制品及食品的安全出现隐患,相关检测部门给予高度的重视。
氟氯戊菊酯及其结构类似物对于人体有不良干扰作用。这些物质主要导致:
⑴损害神经系统;
⑵皮肤黏膜刺激性伤害。
由于烟草成分复杂,致使干扰成分增加,而分析物处于痕量状态,样品前处理(分离待检物)成为安全分析的关键步骤。目前传统的样品前处理技术存在处理时间长、操作繁琐、选择性差等缺点。
技术实现要素:
本发明的目的在于提供一种氟氯戊菊酯分子印迹固相萃取柱的制备方法,克服了现有固相萃取方法的不足,应用于烟叶中氟氯戊菊酯及其结构类似物残留分析中。
本发明技术方案步骤如下:
a、选取氟氰戊菊酯与α-甲基丙烯酸的摩尔配比为1:6;将模板分子氟氰戊菊酯0.1mmol和功能单体α-甲基丙烯酸MAA0.6mmol,放入50mL的安瓿瓶中;
b、安瓿瓶中加入18mL四氢呋喃使其反应,振摇;
c、将在b步骤得到的溶液中再加乙二醇二甲基丙烯酸酯EDMA 8mmol和偶氮二异丁腈AIBN 0.03g;超声1h后,通入N2 脱氧15min后,抽真空1min后密封;
d、将步骤c得到的反应体系在60℃的恒温水浴中静置15h,得块状白色固体氟氰戊菊酯分子印迹聚合物MIP;
e、将步骤d中所得印迹聚合物经研磨、粉碎,过200目筛,再用去离子水沉降聚合物3次除去过细粉末;将最终得到的MIP颗粒用甲醇:乙酸(体积比95:5)洗脱至无模板分子,最后用甲醇浸泡1h除去残留的乙酸,洗脱后的聚合物放入真空干燥器中,40℃条件下,干燥10h,得到具有特异性识别的氟氰戊菊酯分子印迹聚合物MIP;
f、称取步骤e中所制备的分子印迹聚合物MIP100g,溶于水中装入内径为1.5cm的固相萃取空柱内,用固相萃取装置将水抽出后,再用水淋洗柱子,最后在上端再加上少许脱脂棉,轻轻挤压使柱子填充紧实备用,即制备成氟氰戊菊酯分子印迹固相萃取柱。
进一步,所述的致孔剂为四氢呋喃。
进一步,所述的交联剂为乙二醇二甲基丙烯酸酯。
进一步,所述引发剂为偶氮二异丁腈。
一种采用上述方法制备的氟氰戊菊酯分子印迹固相萃取柱在烟叶分析中的应用,具体步骤如下:
a、绘制氟氰戊菊酯的线性关系曲线及检测限
将氟氰戊菊酯分别配制成质量浓度为200、100、50、20、10、5、2、1、0.5μg/mL的一系列标准溶液,绘制氟氰戊菊酯的线性关系曲线;
氟氰戊菊酯的线性关系及检测限
b、新鲜烟叶样品前处理
称取5.000 g新鲜烟叶样品,捣碎,加入6.000 g无水硫酸钠,用以除去样品中过多的水分,加入10 mL四氢呋喃混匀,超声15 min,于4000 r/min离心10 min,收集分离液;再加入10 mL四氢呋喃混匀,超声15 min,于4000 r/min离心10 min,收集分离液与头次分离液合并,过氟氰戊菊酯分子印迹固相萃取柱,用20 mL水淋洗,待排净淋洗液后,用10 mL甲醇洗脱,收集洗脱液留待色谱检测;
c、色谱检测条件
色谱柱:agela C18(4.6×150 mm,5 μm),检测波长200nm,流动相为甲醇-水,甲醇:水体积比为90:10,流速1 mL/min,柱温为25℃,进样量20 µL;
d、分析检测结果
利用利用a步骤绘制的氟氰戊菊酯的性关系曲线线性关系曲线分析色谱检测结果。
本发明采用本体聚合方法制备氟氯戊菊酯分子印迹聚合物,所制备的聚合物经研磨、漂洗、过筛、洗脱、干燥等一系列处理后,再经高效液相色谱法对其进行评价分析即可得到具有特异性吸附效果的聚合物材料,并制备成分子印迹固相萃取柱,用于样品净化,对烟叶中氟氯戊菊酯进行分离和富集,用于对氟氯戊菊酯的检测分析。
本发明的设计思路为:
固相萃取(solid phase extraction,SPE)具有较多优势,如具有高的回收率和高的富集倍数;使用有机溶剂少,减少了对环境的污染;固相分离操作易于收集分析物组分,能处理小体积试样;操作简单快速,易实现自动化。
分子印迹聚合物(Molecularly Imprinted Polymers,MIPs)是指为获得在空间构型和结合位点上与某一目标化合物(模板分子或印迹分子)完全匹配的高分子聚合物,分子印迹技术被形象的比喻为制造识别“分子钥匙”的“人工锁”技术。
MIPs作为SPE的吸附剂可以弥补普通吸附剂选择性差的不足,而且要比免疫吸附剂的稳定性好,还可以重复使用,使痕量被分析物在复杂样品中得到分离富集。
选择分子印迹固相萃取技术是指为获得在空间构型和结合位点上与某一目标化合物(模板分子或印迹分子)完全匹配的高分子聚合物,分子印迹技术被形象的比喻为制造识别“分子钥匙”的“人工锁”技术。
分子印迹固相萃取技术能够克服烟叶样品体系复杂、预处理繁琐等不利因素,达到分离纯化的目的,从而降低检测限,提高分析的精密度和准确性,为痕量组分的富集和分析提供极大的方便。
分子印迹固相萃取技术具有以下优点:(1)对目标物能进行特异性吸附;(2)能耐高温、高压、有机溶剂;(3)重复使用次数多;⑷稳定性好、使用寿命长等优点。它克服了样品体系复杂、预处理繁琐等缺点,因此它已成为分子印迹技术在分析检测中最具应用前景的研究热点。
本发明的有益效果是所提供的分子印迹固相萃取柱可用于烟叶中氟氰戊菊酯以及结构类似物的选择性吸附和富集,适合烟草检测单位、科研单位及卷烟生产企业对产品的监控时使用。与普通的固相萃取柱相比,分子印迹固相萃取柱具有制备过程简单、特异性好、重现性高、分离效果好、回收率高、可反复使用、精密度高等特点。
附图说明
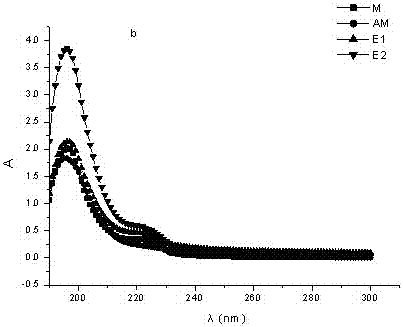
图1氟氰戊菊酯与MAA功能单体紫外吸收光谱;
图2氟氰戊菊酯与AM功能单体紫外吸收光谱;
图3氟氰戊菊酯分子印迹聚合物300nmSEM图;
图4氟氰戊菊酯分子印迹聚合物500nmSEM图;
图5氟氰戊菊酯分子印迹聚合物1.00umSEM图;
图6新鲜烟叶Ⅰ样品洗脱液色谱图;
图7新鲜烟叶Ⅱ样品洗脱液色谱图;
图8氟氰戊菊酯分子印迹固相萃取柱结构示意图;
图9本发明反应中的分子结构图。
图中:M为氟氰戊菊酯;MAA为α-甲基丙烯酸;AM为丙烯酰胺;E1为混合物实际测定吸光值;E2为混合物理论吸光值;氟氰戊菊酯分子印迹聚合物(未洗脱前空穴状态)为具有特异性识别的分子印迹聚合物;氟氰戊菊酯分子印迹聚合物(洗脱后空穴状态)为待重复利用时的具有特异性识别的分子印迹聚合物。
具体实施方式
以下实施例可使专业技术人员全面理解本发明,但不以任何方式限制本发明。
在制备氟氰戊菊酯分子印迹固相萃取柱之前,本发明作了如下前期试验:
(一)功能单体种类的确定
由于混合物在同一波长下紫外吸光值小于模板物质与功能单体分别测定的紫外吸光值的加和(即理论吸光值),可知模板物质与功能单体间发生了相互作用(如图1、图2所示)。
由实验结果可知,氟氰戊菊酯-MAA实测值与理论值差值为1.956,氟氰戊菊酯-AM实测值与理论值差值为1.910。由差值看出氟氰戊菊酯与MAA结合力较大,证明了利用MAA合成的分子印迹聚合物对氟氯戊菊酯具有更好的稳定性和特异识别能力。
(二)不同比例功能单体对MIP吸附能力的影响
合成MIP时,体系内模板物质与功能单体的配比不同其对氟氯戊菊酯的特异性吸附能力的表现也不同,过多或者过少的功能单体都会降低分子印迹聚合物的特异性吸附能力。主要原因是功能单体不足则形成的识别位点少,而过多的功能单体会被无规则固定在交联剂内从而导致非特异性识别位点增加从而降低吸附性能。所以可以通过静态吸附实验来确定最佳配比。
定义印迹因子IF,
IF=QMIP/QNMIP
QMIP——MIP吸附氟氯戊菊酯的量
QNMIP——空白印迹聚合物对氟氯戊菊酯的结合量
IF值越大分子印迹的效果越好。实验结果如下表所示。
表1功能单体加入量对聚合物吸附性能的影响
从表1的实验数据分析得出,模板物质与功能单体MAA的摩尔比为1:6时,聚合的MIP不仅吸附量大而且印迹效果好;造成这个结果的原因为在氟氯戊菊酯与功能单体MAA的摩尔比为1:2和1:4时,ɑ-甲基丙烯酸未与氟氯戊菊酯充分结合,使聚合的MIP吸附量小;而较高的氟氯戊菊酯与功能单体MAA的摩尔比(如1:8)时,过量的ɑ-甲基丙烯酸被固定到MIP内,经研磨暴露出来,导致聚合的MIP非特异性结合点增多,所以印迹效果差。故本实验利用氟氯戊菊酯与功能单体MAA的摩尔比为1:6作为最佳配比聚合。
(三)致孔剂及用量的选择
1.致孔剂选择
1.1化学分析法
称取一定量的氟氰戊菊酯,分别使用乙腈、四氢呋喃和甲醇溶解,配制成0.5mmol/L的溶液,再加入α-甲基丙烯酸(MAA),配制成乙腈为溶剂的氟氰戊菊酯与MAA浓度比1:2、1:4、1:6、1:8、1:10的系列溶液,四氢呋喃与甲醇为致孔剂的溶液配制同上,测定其紫外光谱。实验发现,使用乙腈为致孔剂,溶液最大吸收波长红移了1nm;使用四氢呋喃为致孔剂,溶液最大吸收波长红移了4nm;使用甲醇为致孔剂,溶液最大吸收波长红移了2nm。结果表明四氢呋喃为致孔剂,氟氰戊菊酯与MAA间产生了较强的分子间作用力,所以选择四氢呋喃为致孔剂。
1.2聚合物物理状态分析
乙腈作为致孔剂合成物为乳白色液体,未达到聚合状态;四氢呋喃作为致孔剂合成物为白色多孔固体;甲醇作为致孔剂合成物为黄色透明固体,未达到合成多孔聚合物的目的。所以本实验选择四氢呋喃为致孔剂。
2.致孔剂用量选择
实验通过比对三组致孔剂使用量合成的聚合物状态,确定致孔剂用量。三组聚合物分别加入致孔剂9mL、18mL和27mL,致孔剂加入9mL的聚合物致孔不完全有黄色透明固体;致孔剂加入18mL的聚合物致孔完全且无多余液体;致孔剂加入28mL安瓿瓶中有固体沉淀,震荡后成悬浊液,为未聚合状态。所以选择致孔剂四氢呋喃的用量为18mL。
(四)充N2时间对聚合时间的影响
实验过程中,在热引发聚合前需要在液体内通入氮气,其目的为将液体内部溶解的氧气排除,因为氧自由基导致交联剂链增长终止或按非预期设想的方式增长,且为了避免不受控聚合情况的发生,所以对聚合前体系充入氮气。但是充入氮气时间对聚合时间及聚合物的量有一定的影响。
实验研究了充氮气5 min、15 min以及30 min三种情况,结果发现制作氟氰戊菊酯分子印迹聚合物时,30 min后三个烧瓶内都出现了乳白色聚合物,充氮气15 min和25 min产生的聚合物与反应前溶剂的百分含量呈正相关,充氮气5 min产生的聚合物的量较少,主要原因是充氮气5 min不能将溶液中溶氧完全赶出。在充氮气15 min和25 min条件下,反应5小时后聚合物生成,继续加热聚合物量不再增加。而充氮气25 min产生聚合物的量明显小于充氮气5 min和15 min所产生的聚合物,因为充氮气25 min会使致孔剂大量挥发。所以本实验选择充氮气15 min。
实施例:
氟氰戊菊酯分子印迹聚合物的制备
将氟氰戊菊酯0.1mmol和0.6mmol的α-甲基丙烯酸(MAA),放入50mL的安瓿瓶中,加18mL四氢呋喃使其反应,振摇,再加交联剂乙二醇二甲基丙烯酸酯(EDMA) 8mmol和引发剂偶氮二异丁腈(AIBN) 0.03g。超声1h后通入N2 脱氧15min后,抽真空1min后密封,在60℃的恒温水浴中静置15h,得块状白色固体MIP。经研磨、粉碎,过200目筛,再用去离子水沉降聚合物3次除去过细粉末。将最终得到的MIP颗粒用甲醇:乙酸(体积比95:5)洗脱至无模板分子,最后用甲醇浸泡1h除去残留的乙酸,洗脱后的聚合物放入真空干燥器中(40℃)干燥10h,得到氟氰戊菊酯分子印迹聚合物MIP,用发射场扫描电镜观察其外观,如图3-图5所示。图9表示本发明反应中的分子结构变化。
分子印迹固相萃取柱的制备
称取100mg的氟氰戊菊酯分子印迹聚合物MIP,溶于水中装入内径为1.5cm的固相萃取空柱内,用固相萃取装置将水抽出后,再用水淋洗柱子,最后在上端再加上少许脱脂棉,轻轻挤压使柱子填充紧实备用。固相萃取柱如图8所示。
氟氰戊菊酯分子印迹固相萃取柱在新鲜烟叶检测中应用
a、样品前处理
分别称取5.000 g样品(新鲜烟叶),捣碎,加6.000 g无水硫酸钠(用以除去样品中过多的水分),用20 mL四氢呋喃浸溶,并提取两次,每次10 mL,混匀;超声15 min,于4000 r/min离心10 min,吸取上清液过分子印迹固相萃取柱,用20 mL水淋洗,待排净淋洗液后,用10 mL甲醇洗脱,收集洗脱液待检。
b、线性关系与检测限
将氟氰戊菊酯分别配制成质量浓度为200,100,50,20,10,5,2,1,0.5μg/mL的一系列标准溶液,绘制氟氰戊菊酯的线性关系曲线;其线性关系与检测限结果如表2所示:
表2 氟氰戊菊酯的线性关系及检测限
应是用线性关系,分析新鲜烟叶Ⅰ,新鲜烟叶Ⅱ中的氟氰戊菊酯含量。
c、回收率和精密度试验
采用本方法对新鲜烟叶Ⅰ,新鲜烟叶Ⅱ,在1 μg/g和100μg/g 2个添加水平下,进行添加回收率试验,分析结果如表3所示,平均回收率在96.7%~99.9%之间,相对标准偏差(RSD)在0.93%~2.18%之间(n=5)。说明本发明方法的回收率和精密度良好。
表3 新鲜烟叶Ⅰ、新鲜烟叶Ⅱ的回收率和精密度试验(n=5)
以上借助较佳的实施例对本发明技术方案进行的详细说明是示意性的而非形式上的限制。本领域的技术人员在阅读本发明说明书的基础上,可以对实施例所记载的技术方案进行修改,或者对其中部分技术特征进行等同替换,而这些修改或者替换,并不使相应技术方案的本质脱离本发明实施例技术方案的范围。
- 还没有人留言评论。精彩留言会获得点赞!