一种高活性催化材料及其制备方法与流程
本发明涉及一种催化材料及其制备方法,更进一步说本发明涉及一种具有拟薄水铝石结构的催化材料及其制备方法。
背景技术
流化催化裂化作为石油炼制加工过程中的重要反应过程,在石油加工工业中得到广泛应用,在炼油厂中占有举足轻重的地位。在催化裂化反应过程中,重质馏分如减压馏分油或更重组分的渣油在催化剂存在下发生裂化反应,转化为汽油、柴油等液态裂化产品以及较轻的气态裂化产品,催化裂化反应一般遵循正碳离子反应机理,因此在反应过程中通常需要使用具有较高酸性特别是具有b酸中心的催化材料。
在早期的催化裂化催化剂中曾以无定形硅铝材料作为一种活性组分,它不具有典型的晶体结构,但同时含有b酸中心和l酸中心,也是一类酸性材料,但该材料的水热结构稳定性较差,裂化活性相对较低而且需要的反应温度较高,因此逐渐被稳定性好、酸性强的结晶分子筛所替代。沸石分子筛是一类具有规整孔道结构、较强酸性以及较好稳定性的催化材料,在裂化反应中表现出很好的催化反应性能,因此在石油炼制和加工工业中得到广泛应用。伴随着经济的发展石油资源正面临着日益耗竭的局面,原油的重质化、劣质化趋势也在不断加剧,掺渣比例不断提高,因此近年来更加重视对重油和渣油的深加工,部分炼厂已开始掺炼减压渣油,甚至直接以常压渣油作为裂化反应原料来使用。微孔沸石分子筛的孔道相对较小,一般情况下孔道尺寸小于2nm,对于重油或渣油等大分子而言分子筛的孔道限制作用较为明显,因此对大分子的裂化能力稍显不足。
介孔材料的研制开发为提高大分子反应性能提供了可能。介孔材料是一类孔径介于2~50nm的多孔材料,也称中孔材料,其孔道尺寸非常适合重油等大分子的裂化反应,因此催化领域的研究人员在介孔材料的开发中投入了极大的兴趣。介孔材料又可分为有序介孔材料和无序介孔材料,有序介孔材料多指长程有序短程无序的介孔材料,其孔道尺寸均匀,孔径分布窄,孔道排列可呈现为一维、二维或三维孔道分布情况,即长程有序,但其孔壁为非晶体结构,即短程无序,也有部分材料是完全无序连接的,呈蠕虫状排列,三维互通;无序介孔材料无论在长程还是短程上均为无序结构,其孔道尺寸不均一,孔分布较宽,孔道的连接也是无序的。在有序介孔材料的制备过程中通常需要使用模板剂,如表面活性剂、高分子嵌段共聚物等,制备成本相应的会有所提高,部分模板剂对环境不友好,这在一定程度上阻碍了有序介孔材料的工业应用,特别是在催化裂化反应过程中的应用。而无序介孔材料的制备过程中基本不需要使用模板剂,制备成本大幅降低,因此对实际应用而言更多的研究工作集中于无序介孔材料的开发。
us5,051,385中公开了一种单分散的中孔硅铝复合材料,通过酸性无机铝盐和硅溶胶的混合再加入碱进行反应而制得,其铝含量约5~40重量%,孔径为20~50nm,比表面积为50~100m2/g。
us4,708,945中公开了一种硅铝材料,是先在多孔一水软铝石上负载氧化硅粒子或水合氧化硅,再将所得复合物在600℃以上水热处理一定时间,其中氧化硅与过渡态一水软铝石的羟基相结合,所得材料的比表面积达100~200m2/g,平均孔径为7~7.5nm。
us4,440,872中公开了一系列酸性裂化催化剂,其中一些催化剂的载体是通过在γ-al2o3上浸渍硅烷,然后经500℃焙烧或水蒸汽处理后制得的。
us2,394,796中公开了一种复合材料,在多孔水合氧化铝上浸渍四氯化硅或四乙基硅,然后经水解获得硅铝复合材料。
cn1353008a中公开了一种硅铝催化材料,采用无机铝盐和水玻璃为原料,经沉淀、洗涤、解胶等过程形成稳定的硅铝溶胶,经干燥得到白色凝胶,再于350℃~650℃下焙烧1~20小时得到硅铝催化材料。
cn1565733a中公开了一种硅铝材料,该材料具有拟薄水铝石结构,孔径分布集中,比表面积约200~400m2/g,孔容为0.5~2.0ml/g,平均孔径为8~20nm,最可几孔径为5~15nm。其制备方法是将铝源与碱溶液在室温至85℃下中和成胶,成胶终点ph为7~11;然后按照sio2∶al2o3=1∶(0.6-9)的重量比加入硅源,在室温至90℃下老化1~10小时;将所得固体沉淀物进行铵交换除去杂质离子;再在100℃~150℃下干燥、350℃~650℃下焙烧1~20小时。
技术实现要素:
本发明的目的之一是提供有别于现有技术、具有独特的结构特点、b酸中心比例和铝分布特性的催化材料。
本发明的目的之二是提供一种相应的制备方法。
本发明的目的之三是提供该催化材料的应用。
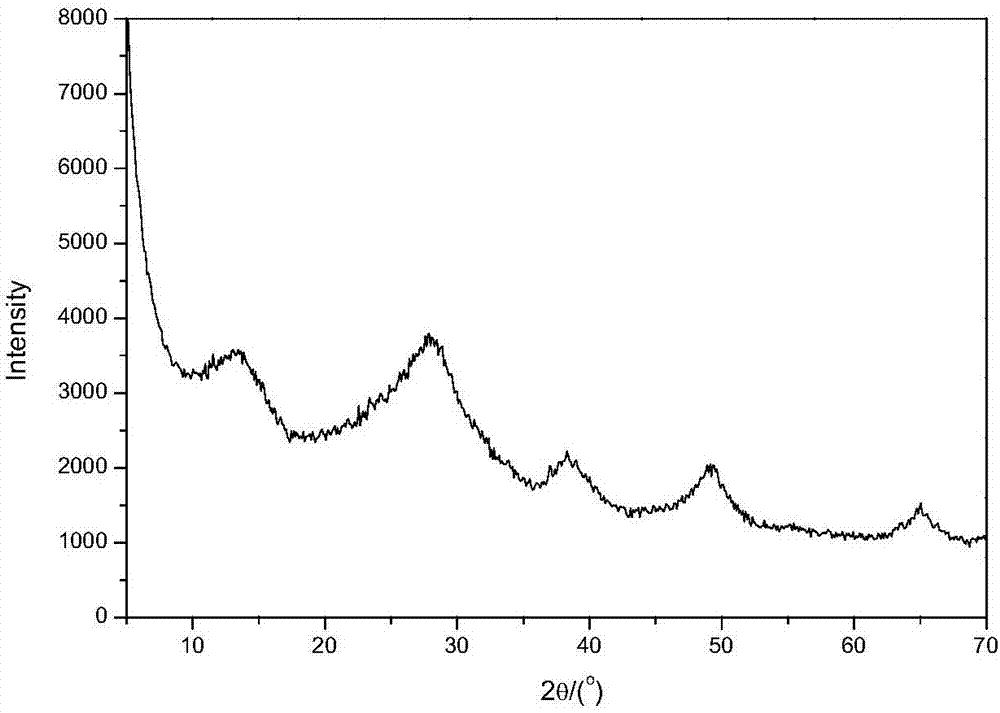
在本发明的第一个方面中,所提供的催化材料,其特征在于xrd谱图在2θ角为14°、28°、38.5°、49°和65°处存在拟薄水铝石结构的特征衍射峰,200℃下吡啶红外测得的b酸中心数量与l酸中心数量的比值为0.10~0.23,以氧化物重量计的化学组成中含有15~45%的硅和55~85%的铝,比表面积为300~500m2/g,平均孔径为5~18nm,当c为xps方法测得的表面al/si原子比值,d为xrf方法测得的体相al/si原子比值时,c/d=1.2~1.6。
本发明的催化材料,比表面积为300~500m2/g、优选320~480m2/g;平均孔径5~18nm、优选6~15nm。
本发明的催化材料,分别通过xps方法和xrf方法进行元素含量的表征。x射线光电子能谱(xps)主要用于材料表面纳米级深度元素组成及分布情况的表征。分析使用的激发源为单色化的功率150w的alkαx射线,荷电位移用来自污染碳的c1s峰(284.8ev)校正,根据al2p的原子含量和si2p的原子含量来计算材料表面的al/si原子比值。x射线荧光光谱(xrf)主要用于对材料体相化学组成进行分析,根据所测al和si的含量计算出体相al/si原子比。。当xps方法测得的表面al/si原子比值为c,xrf方法测得的体相al/si原子比值为d时,所述的c/d=1.2~1.6、优选1.25~1.56。
本发明的催化材料,所说的b(bronsted)酸中心数量与l(lewis)酸中心数量是采用吡啶红外光谱法获得的。所述的吡啶红外光谱法是将催化材料样品自撑压片,置于红外光谱仪的原位池中密封,升温至350℃并抽真空至10-3pa,恒温1小时后脱除样品吸附的气体分子;冷却至室温后导入吡啶蒸气保持吸附平衡30分钟,然后升温至200℃,重新抽真空至10-3pa并在此真空度下脱附30分钟,降至室温摄谱,扫描范围1400~1700cm-1,即可获得样品经200℃脱附的吡啶吸附红外光谱图。根据吡啶吸附红外光谱图中1540cm-1和1450cm-1特征吸收峰的强度,计算b酸中心与l酸中心的相对量。所述的200℃下吡啶红外测得的b酸中心数量与l酸中心数量的比值为0.10~0.23、优选为0.120~0.215。
在本发明的第二个方面中,还提供了上述催化材料的制备方法,在室温至60℃及搅拌下以并流方式同时将硅源和碱性铝源加入到容器中控制ph值为13~14进行混合成胶,然后将酸性铝源加入其中,并控制浆液体系的终点ph值为8.0~10.5,然后在40~80℃温度下恒温处理、如1~8小时,洗涤、过滤,将所得固体沉淀物进行离子交换除去杂质离子,其中,硅源与铝源的重量比例1:(1.2~5.7)、、优选1:(1.3~4.3),硅源以氧化硅计,铝源以氧化铝计、为所述碱性铝源和所述酸性铝源之和,。
所说的制备方法中,硅源选自水玻璃、硅酸钠、碱性硅溶胶、四乙氧基硅和四甲氧基硅中的一种或多种。
所说的制备方法中,碱性铝源为偏铝酸钠,可以为不同苛性比、不同浓度的偏铝酸钠。所述的苛性比优选1.5~11.5、更优选1.65~2.55,所述的浓度优选40~200gal2o3/l、更优选41~190gal2o3/l。
所说的制备方法中,加入物料的并流方式的概念为本领域技术人员所知晓,是指将n+1(n≥1)种物料(如本发明中的硅源和碱性铝源两种物料)同时向容器中加入进行混合,使得每种物料保持匀速加入、n+1种物料均在相同的时间内加入完成的操作方式。例如,具体的操作中可使用蠕动泵,控制分别用于输送硅源和碱性铝源的蠕动泵的单位时间内的流量参数,并匀速进行以保证硅源和碱性铝源这两种物料在相同的时间内加完。
所说的制备方法中,酸性铝源选自硝酸铝、硫酸铝和氯化铝中的一种或多种。
所说的制备方法中,所说的离子交换除去杂质离子的过程,是指铵交换过程,是将恒温处理后过滤得到的固体沉淀物按沉淀物干基:铵盐:h2o=1:(0.2~1):(10~30)的重量比在室温至100℃下交换,交换时间0.5~1小时,可重复多次交换,直至固体沉淀物中氧化钠含量低于0.3%;所说的铵交换过程中,铵盐选自氯化铵、硫酸铵、硝酸铵、碳酸铵和碳酸氢铵中的一种或多种。
本发明提供的活性催化材料,具有拟薄水铝石结构,表面性质特殊,表面al/si原子比明显高于体相al/si原子比,介孔特征明显,b酸中心比例高,重油转化能力强,液体收率高。因此,在本发明的第三个方面中,还提供了催化材料的应用,例如在石油化工过程作为重油转化的催化剂活性组分或基质的应用。
附图说明
图1为本发明的催化材料的x射线衍射谱图。
图2为本发明的催化材料的低温氮气吸脱附等温曲线。
图3为本发明的催化材料的bjh孔径分布曲线。
具体实施方式
下面的实施例将对本发明作进一步的说明,但并不因此而限制本发明。
在各实施例中,样品的na2o、al2o3、sio2含量用x射线荧光法(xrf)测定(参见《石油化工分析方法(ripp实验方法)》,杨翠定等编,科学出版社,1990年出版)。样品表面的al、si原子含量用x射线光电子能谱(xps)测定。样品的物相采用x射线衍射法测定。bet比表面、孔结构等物化数据采用低温氮吸附-脱附法测定。样品的酸性数据采用红外吡啶吸附原位测量法测定。
实施例1
本实例说明本发明提供的催化材料及其制备过程。
在烧杯中加入少量的去离子水,在室温及剧烈搅拌下以并流方式将四乙氧基硅和偏铝酸钠溶液(160gal2o3/l,苛性比2.55)同时加入到烧杯中进行混合成胶,保证两种物料在相同的时间内加完,ph值为13.4;然后将al(no3)3溶液(浓度60gal2o3/l)加入其中并控制浆液体系的终点ph值为10.2,随后在80℃下恒温处理2小时;洗涤过滤后将所得固体沉淀物按沉淀物干基:铵盐:h2o=1:0.6:10的重量比在60℃下交换0.5小时,过滤、水洗得到的催化材料记为fab-1。
fab-1的x射线衍射谱图如图1所示,其中在2θ角为14°、28°、38.5°、49°和65°处出现5个特征衍射峰,为典型的拟薄水铝石结构;其低温氮气吸脱附等温曲线如图2所示,呈现iv类等温线形式,有滞后环的存在,说明材料具有典型的介孔特征,其bet比表面积为351m2/g;其bjh孔径分布曲线如图3所示,平均孔径为15nm。
fab-1由xrf方法测得的化学组成为:0.09na2o·39.2sio2·59.8al2o3,则由xrf方法测得的体相al/si原子比d为1.73;由xps方法测得的表面al/si原子比c为2.33,因此c/d=1.35。
fab-1在200℃下吡啶红外测得的b酸中心数量与l酸中心数量的比值为0.189。
实施例2
本实例说明本发明提供的催化材料及其制备过程。
在烧杯中加入少量的去离子水,在40℃及剧烈搅拌下以并流方式将水玻璃溶液(浓度100gsio2/l)和偏铝酸钠溶液(160gal2o3/l,苛性比2.55)同时加入到烧杯中进行混合成胶,保证两种物料在相同的时间内加完,ph值为13.7;然后将al2(so4)3溶液(浓度60gal2o3/l)加入其中并控制浆液体系的终点ph值为9.3,随后在40℃下恒温处理4小时;洗涤过滤后将所得固体沉淀物按沉淀物干基:铵盐:h2o=1:0.8:12的重量比在60℃下交换1小时,过滤、水洗得到的催化材料记为fab-2。
fab-2的x射线衍射谱图具有图1所示特征,具有典型的拟薄水铝石结构;其低温氮气吸脱附等温曲线具有图2所示特征,具有典型的介孔特征,其bet比表面积为428m2/g;其bjh孔径分布曲线具有图3所示特征,平均孔径为9nm。
fab-2由xrf方法测得的化学组成为:0.18na2o·22.5sio2·77.0al2o3,则由xrf方法测得的体相al/si原子比d为3.88;由xps方法测得的表面al/si原子比c为5.84,c/d=1.50。
fab-2在200℃下吡啶红外测得的b酸中心数量与l酸中心数量的比值为0.127。
实施例3
本实例说明本发明提供的催化材料及其制备过程。
在烧杯中加入少量的去离子水,在50℃及剧烈搅拌下以并流方式将碱性硅溶胶(浓度21gsio2/l)和偏铝酸钠溶液(102gal2o3/l,苛性比2.45)同时加入到烧杯中进行混合成胶,保证两种物料在相同的时间内加完,ph值为13.3;然后将al2(so4)3溶液(浓度60gal2o3/l)加入其中并控制浆液体系的终点ph值为10.0,随后在70℃下恒温处理6小时;洗涤过滤后将所得固体沉淀物按沉淀物干基:铵盐:h2o=1:0.4:10的重量比在50℃下交换0.5小时,过滤水洗后,重复一次铵交换过程,过滤、水洗得到的催化材料记为fab-3。
fab-3的x射线衍射谱图具有图1所示特征,具有典型的拟薄水铝石结构;其低温氮气吸脱附等温曲线具有图2所示特征,具有典型的介孔特征,其bet比表面积为386m2/g;其bjh孔径分布曲线具有图3所示特征,平均孔径为13nm。
fab-3由xrf方法测得的化学组成为:0.12na2o·35.0sio2·64.3al2o3,则由xrf方法测得的体相al/si原子比d为2.08;由xps方法测得的表面al/si原子比c为2.89,c/d=1.39。
fab-3在200℃下吡啶红外测得的b酸中心数量与l酸中心数量的比值为0.175。
对比例1
本对比例说明采用非并流加入方式所制备得到的对比催化材料。
取一定量的碱性硅溶胶(浓度21gsio2/l)置于烧杯中,升温至55℃,然后在搅拌下加入定量的偏铝酸钠溶液(102gal2o3/l,苛性比2.45),ph值为13.3,搅拌10分钟后,在剧烈搅拌下加入al2(so4)3溶液(浓度60gal2o3/l),并调节浆液终点ph值为10.0,随后在70℃下恒温处理6小时;洗涤过滤后将所得固体沉淀物按沉淀物干基:铵盐:h2o=1:0.4:10的重量比在50℃下交换0.5小时,过滤水洗后,重复一次铵交换过程,过滤后得到对比的催化材料,记为db-1。
db-1的x射线衍射谱图具有图1所示特征,具有典型的拟薄水铝石结构;其bet比表面积为390m2/g,平均孔径为13nm,bjh孔径分布曲线具有图3所示特征。
db-1由xrf方法测得的化学组成为:0.19na2o·34.8sio2·64.6al2o3,则由xrf方法测得的体相al/si原子比d为2.11;由xps方法测得的表面al/si原子比c为3.69,c/d=1.75。db-2在200℃下吡啶红外测得的b酸中心数量与l酸中心数量的比值为0.146。
实施例4
本实例说明本发明提供的催化材料及其制备过程。
在烧杯中加入少量的去离子水,在35℃及剧烈搅拌下以并流方式将水玻璃溶液和偏铝酸钠溶液(41gal2o3/l,苛性比11.5)同时加入到烧杯中进行混合成胶,保证两种物料在相同的时间内加完,ph值为13.7;然后将alcl3溶液(浓度60gal2o3/l)加入其中并控制浆液体系的终点ph值为8.8,随后在50℃下恒温处理8小时;洗涤过滤后将所得固体沉淀物按沉淀物干基:铵盐:h2o=1:0.7:12的重量比在50℃下交换0.5小时,过滤水洗后,重复一次铵交换过程,过滤、水洗得到的催化材料记为fab-4。
fab-4的x射线衍射谱图具有图1所示特征,具有典型的拟薄水铝石结构;其低温氮气吸脱附等温曲线具有图2所示特征,具有典型的介孔特征,其bet比表面积为319m2/g;其bjh孔径分布曲线具有图3所示特征,平均孔径为17nm。
fab-4由xrf方法测得的化学组成为:0.25na2o·43.5sio2·56.0al2o3,则由xrf方法测得的体相al/si原子比d为1.46;由xps方法测得的表面al/si原子比c为1.85,c/d=1.27。
fab-4在200℃下吡啶红外测得的b酸中心数量与l酸中心数量的比值为0.211。
对比例2
本对比例说明采用非并流加入方式所制备得到的对比催化材料。
取一定量的水玻璃溶液置于烧杯中,在35℃及搅拌下加入定量的偏铝酸钠溶液(41gal2o3/l,苛性比11.5),ph值为13.7;搅拌10分钟后,在剧烈搅拌下加入alcl3溶液(浓度60gal2o3/l),并调节浆液终点ph值为8.8,随后在50℃下恒温处理8小时;洗涤过滤后将所得固体沉淀物按沉淀物干基:铵盐:h2o=1:0.7:12的重量比在50℃下交换0.5小时,过滤水洗后,重复一次铵交换过程,过滤后得到对比的催化材料,记为db-2。
db-2的x射线衍射谱图具有图1所示特征,具有典型的拟薄水铝石结构;其bet比表面积为327m2/g,平均孔径为16nm,bjh孔径分布曲线具有图3所示特征。
db-2由xrf方法测得的化学组成为:0.20na2o·43.8sio2·55.8al2o3,则由xrf方法测得的体相al/si原子比d为1.44;由xps方法测得的表面al/si原子比c为2.2,c/d=1.53。db-2在200℃下吡啶红外测得的b酸中心数量与l酸中心数量的比值为0.169。
实施例5
本实例说明本发明提供的催化材料及其制备过程。
在烧杯中加入少量的去离子水,在60℃及剧烈搅拌下以并流方式将四乙氧基硅和偏铝酸钠溶液(102gal2o3/l,苛性比2.45)同时加入到烧杯中进行混合成胶,保证两种物料在相同的时间内加完,ph值为13.4;然后将alcl3溶液(浓度60gal2o3/l)加入其中并控制浆液体系的终点ph值为9.0,随后在80℃下恒温处理4小时;洗涤过滤后将所得固体沉淀物按沉淀物干基:铵盐:h2o=1:0.6:15的重量比在70℃下交换0.5小时,过滤、水洗得到的催化材料记为fab-5。
fab-5的x射线衍射谱图具有图1所示特征,具有典型的拟薄水铝石结构;其低温氮气吸脱附等温曲线具有图2所示特征,具有典型的介孔特征,其bet比表面积为420m2/g;其bjh孔径分布曲线具有图3所示特征,平均孔径为11nm。
fab-5由xrf方法测得的化学组成为:0.10na2o·28.6sio2·70.8al2o3,则由xrf方法测得的体相al/si原子比d为2.81;由xps方法测得的表面al/si原子比c为4.01,c/d=1.43。
fab-5在200℃下吡啶红外测得的b酸中心数量与l酸中心数量的比值为0.141。
实施例6
本实例说明本发明提供的催化材料及其制备过程。
在烧杯中加入少量的去离子水,在30℃及剧烈搅拌下以并流方式将水玻璃溶液和偏铝酸钠溶液(180gal2o3/l,苛性比1.65)同时加入到烧杯中进行混合成胶,保证两种物料在相同的时间内加完,ph值为13.8;然后将al(no3)3溶液(浓度60gal2o3/l)加入其中并控制浆液体系的终点ph值为8.5,随后在60℃下恒温处理4小时;洗涤过滤后将所得固体沉淀物按沉淀物干基:铵盐:h2o=1:0.8:10的重量比在55℃下交换1小时,过滤、水洗得到的催化材料记为fab-6。
fab-6的x射线衍射谱图具有图1所示特征,具有典型的拟薄水铝石结构;其低温氮气吸脱附等温曲线具有图2所示特征,具有典型的介孔特征,其bet比表面积为463m2/g;其bjh孔径分布曲线具有图3所示特征,平均孔径为7nm。
fab-6由xrf方法测得的化学组成为:0.15na2o·19.1sio2·80.7al2o3,则由xrf方法测得的体相al/si原子比d为4.79;由xps方法测得的表面al/si原子比c为7.47,c/d=1.56。
fab-6在200℃下吡啶红外测得的b酸中心数量与l酸中心数量的比值为0.120。
对比例3
本对比例说明采用非并流加入方式所制备得到的对比催化材料。
取一定量的水玻璃溶液(浓度100gsio2/l)置于烧杯中,升温至60℃,然后在搅拌下加入定量的偏铝酸钠溶液(180gal2o3/l,苛性比1.65),ph值为13.8,搅拌10分钟后,在剧烈搅拌下加入al(no3)3溶液,并调节浆液终点ph值至8.5,然后升温至60℃并恒温处理4小时;洗涤过滤后将所得固体沉淀物按沉淀物干基:铵盐:h2o=1:0.8:10的重量比在55℃下交换1小时,过滤水洗后得到对比的催化材料,记为db-3。
db-3的x射线衍射谱图具有图1所示特征,具有典型的拟薄水铝石结构;其bet比表面积为457m2/g,平均孔径为7nm,bjh孔径分布曲线具有图3所示特征。
db-3由xrf方法测得的化学组成为:0.23na2o·18.5sio2·81.0al2o3,则由xrf方法测得的体相al/si原子比d为5.15;由xps方法测得的表面al/si原子比c为9.30,c/d=1.806。db-3在200℃下吡啶红外测得的b酸中心数量与l酸中心数量的比值为0.093。
实施例7
本实例说明本发明提供的催化材料及其制备过程。
在烧杯中加入少量的去离子水,在45℃及剧烈搅拌下以并流方式将碱性硅溶胶和偏铝酸钠溶液(180gal2o3/l,苛性比1.65)同时加入到烧杯中进行混合成胶,保证两种物料在相同的时间内加完,ph值为13.6;然后将alcl3溶液(浓度60gal2o3/l)加入其中并控制浆液体系的终点ph值为9.7,随后在70℃下恒温处理5小时;洗涤过滤后将所得固体沉淀物按沉淀物干基:铵盐:h2o=1:0.3:10的重量比在70℃下交换0.5小时,过滤水洗后,重复一次铵交换过程,过滤、水洗得到的催化材料记为fab-7。
fab-7的x射线衍射谱图具有图1所示特征,具有典型的拟薄水铝石结构;其低温氮气吸脱附等温曲线具有图2所示特征,具有典型的介孔特征,其bet比表面积为404m2/g;其bjh孔径分布曲线具有图3所示特征,平均孔径为12nm。
fab-7由xrf方法测得的化学组成为:0.11na2o·30.9sio2·68.2al2o3,则由xrf方法测得的体相al/si原子比d为2.50;由xps方法测得的表面al/si原子比c为3.70,c/d=1.48。
fab-7在200℃下吡啶红外测得的b酸中心数量与l酸中心数量的比值为0.162。
实施例8
本实例说明本发明提供的催化材料用于重油裂化反应的性能。
将上述实施例1~7得到的样品fab-1~fab-7样品分别与rehy分子筛按重量比1:4的比例混合均匀,压片后筛分成20~40目颗粒,在800℃、100%水蒸气条件下老化处理12小时,然后在重油微反评价装置上进行反应活性的测试。
原料油性质列于表1。评价结果列于表2。
所述对比样品db-1、db-2和db-3分别与本发明实施例3样品fab-3、实施例4样品fab-4、实施例6样品fab-6的组成相当,作为对比,将对比例1、2、3得到的对比样品db-1、db-2、db-3分别与rehy分子筛按重量比1:4的比例混合均匀,压片后筛分成20~40目颗粒,在800℃、100%水蒸气条件下老化处理12小时,然后在重油微反评价装置上进行反应活性的测试。
评价结果列于表3,表3中同时列出组成相当的本发明样品fab-3、fab-4、fab-6的活性测试结果。
重油微反评价条件:原料油为武混三,样品装量2g,剂油比1.45,反应温度500℃,再生温度600℃。
表1
表2
由表2数据可见,将实施例中的催化材料fab-1~fab-7与rehy分子筛混合并经过高温水热老化处理后,在重油裂化反应中显示出很好的裂化活性,转化率达到69.09%~71.07%,重油收率仅为9.97%~11.11%,重油与焦炭的比为0.995~1.168,表明重油转化能力强,总液体收率高达77.45%~78.40%。
表3
由表3数据可以发现,在与化学组成相当、采用非并流法制备的对比材料db-1~db-3进行比较时,采用本发明方法得到的催化材料具有更高的裂化活性,重油转化能力更强,焦炭产率更低,焦炭的选择性更好,产品分布得到进一步优化。由于本发明得到的催化材料表面富铝程度降低,硅与铝得到更有效的结合,促进了b酸中心比例的提高,因此进一步提高了在大分子裂化反应中的裂化能力。
- 还没有人留言评论。精彩留言会获得点赞!