一种双单原子助剂负载型氧化铜催化剂、其制备方法及用途与流程
本发明属于催化剂技术领域,涉及一种双单原子助剂负载型氧化铜催化剂、其制备方法及用途。
背景技术:
单原子催化剂是多相催化领域一个新兴的研究热点,由于单原子催化剂的活性组分可以最大程度分散,具有最大化的原子利用率,同时电子结构独特,组成和结构单一,可避免因活性组分组成和结构不均匀导致的副反应,在许多反应中表现出比传统纳米粒子更优异的催化活性和选择性,引起了人们极大的研究兴趣。然而单原子催化剂极易聚集,尤其在真实工业反应条件下(一般为高温高压),如何提高单原子催化剂的稳定性是研究的难点之一。
目前可以通过载体本征的一些缺陷作为锚定位来解决单原子催化剂稳定性的问题,但大部分的研究工作集中于载体的表面空位或官能团来对其锚定。cn107096536a公开了一种非贵金属单原子催化剂的可控制备方法,先在多孔载体材料上制得含氧官能团,然后非贵金属离子与含氧官能团发生定点反应,负载到多孔材料上,最后用还原剂还原金属阳离子,得到单原子催化剂,但上述单原子催化剂中单原子一般作为活性组分,且该活性组分通过载体上的含氧官能团进行固定,应用时主要是单原子活性组分起作用,载体所起作用有限。
现有的催化反应中,助剂往往与催化剂共同使用,助剂尽管用量较少,但可以显著提升催化剂的性能,在工业上非常重要。目前,助剂的加入主要是通过外加掺混,分散不均匀,分散度低,通常以纳米粒子的形式存在,原子利用率也较低。虽然现有技术中也有改进研究,cn104148120a公开了一种使用助剂控制催化剂金属中心高度分散的方法,但该方法中助剂需要先进入特定的前体中,适用范围较窄,主要目的是实现贵金属的分散,其自身的对催化性能的提升未有提及。因此,将单原子性能好、结构单一的优势应用于助剂,不仅有助于阐明助剂在催化反应中的作用机理,而且有可能为催化剂的性能优化开辟新路径。现有研究已表明将助剂设计为单原子,可以推动主催化剂表面空位的形成,从而促进催化性能的提升(liangwangetal.,nat.commun.2018,9,1362;abhayadatyeetal.,j.am.chem.soc.2018,140,12964-1297)。工业中助剂的使用往往在两种以上,探索双单原子助剂的制备以及它们之间的协同作用对催化反应过程的影响,对工业催化具有重要的意义。
综上所述,实现双单原子助剂负载型催化剂的稳定合成具有重要的研究价值,是目前催化剂研究的重要方向之一。
技术实现要素:
针对现有技术存在的问题,本发明的目的在于提供一种双单原子助剂负载型氧化铜催化剂、其制备方法及用途,所述催化剂充分利用单原子性能好、结构单一的优势,将单原子作为助剂,负载在主催化剂上,通过单原子之间以及单原子与主催化剂之间的相互作用,增强催化剂的稳定性与催化活性。
为达此目的,本发明采用以下技术方案:
第一方面,本发明提供了一种双单原子助剂负载型氧化铜催化剂,所述催化剂包括主催化剂cuo和单原子助剂sn和zn,所述单原子助剂负载在主催化剂表面上。
本发明中,所述催化剂以氧化铜为主体,锡和锌以单原子的形式进入氧化铜表面的晶格或空位,分散性较好,稳定性强,而且两者作为助剂而非主要活性组份,使用量极大的减少,却能利用单原子的优势,通过双单原子之间以及其与氧化铜之间的相互作用,显著提高用于催化反应时的催化活性,并提高相应产物的选择性。
以下作为本发明优选的技术方案,但不作为本发明提供的技术方案的限制,通过以下技术方案,可以更好地达到和实现本发明的技术目的和有益效果。
作为本发明优选的技术方案,所述催化剂中cuo的质量百分含量为98.00~99.99wt%,例如98.00wt%、98.20wt%、98.40wt%、98.60wt%、98.80wt%、99.00wt%、99.20wt%、99.40wt%、99.60wt%、99.80wt%或99.99wt%等,但并不仅限于所列举的数值,该数值范围内其他未列举的数值同样适用。
优选地,所述催化剂中sn和zn的质量百分含量独立地为0.005~1.00wt%,例如0.005wt%、0.01wt%、0.05wt%、0.10wt%、0.20wt%、0.30wt%、0.50wt%、0.70wt%、0.90wt%或1.00wt%等,但并不仅限于所列举的数值,该数值范围内其他未列举的数值同样适用。
本发明中,sn和zn作为助剂负载在主催化剂上,其含量较低,有助于其以单元子形式分散,若其负载量偏高,会使得sn和zn以纳米粒子的形式负载,无法充分发挥单原子结构和性能方面的优势,与主催化剂之间的相互作用反而减弱,不利于催化活性的提高。
优选地,所述单原子助剂sn和zn在催化剂表面分布均一。
优选地,所述催化剂中sn和zn均以单原子分散的状态存在。
优选地,所述催化剂呈片状。
优选地,所述催化剂的厚度为50~60nm,例如50nm、52nm、54nm、55nm、56nm、58nm或60nm等,但并不仅限于所列举的数值,该数值范围内其他未列举的数值同样适用。
第二方面,本发明提供了一种上述双单原子助剂负载型氧化铜催化剂的制备方法,所述方法包括如下步骤:
(1)向铜前驱体和锡前驱体混合溶液中加入碱液,得到悬浊液;
(2)将步骤(1)得到的悬浊液老化后进行水热反应,所得产物固液分离,得到第一固体产物;
(3)向步骤(2)得到的第一固体产物的分散液中加入锌前驱体溶液,固液分离得到第二固体产物;
(4)将步骤(3)得到的第二固体产物焙烧处理,得到双单原子助剂负载型氧化铜催化剂。
本发明中,所述制备方法主要分为两个阶段,首先采用水热法向cuo晶格中引入单原子助剂sn,与此同时,cuo表面产生了大量的铜离子空位,然后采用浸渍-焙烧法将单原子助剂zn锚定在铜离子空位上,得到的双单原子助剂负载的催化剂,所述催化剂中单原子与氧化铜的结合作用较强,具有较高的稳定性。
作为本发明优选的技术方案,步骤(1)所述混合溶液中铜离子的浓度为0.26~1.60mol/l,例如0.26mol/l、0.40mol/l、0.60mol/l、0.80mol/l、1.00mol/l、1.20mol/l、1.40mol/l或1.60mol/l等,但并不仅限于所列举的数值,该数值范围内其他未列举的数值同样适用。
优选地,步骤(1)所述混合溶液中铜离子与锡离子的摩尔比为(1500~2100):1,例如1500:1、1600:1、1700:1、1800:1、1900:1、2000:1或2100:1等,但并不仅限于所列举的数值,该数值范围内其他未列举的数值同样适用。
优选地,步骤(1)所述铜前驱体为可溶性铜盐,锡前驱体为可溶性锡盐。
优选地,所述可溶性铜盐包括硝酸铜、硫酸铜、氯化铜或醋酸铜中的任意一种或至少两种的组合,所述组合典型但非限制性实例有:硝酸铜和氯化铜的组合,硝酸铜和醋酸铜的组合,硫酸铜和醋酸铜的组合,硝酸铜、氯化铜和醋酸铜的组合,硫酸铜、氯化铜和醋酸铜的组合等。
优选地,所述可溶性锡盐包括硝酸锡、硫酸锡、氯化锡或醋酸锡中的任一种或至少两种的组合,所述组合典型但非限制性实例有:硝酸锡和氯化锡的组合,硝酸锡和醋酸锡的组合,硫酸锡和氯化锡的组合,硝酸锡、氯化锡和醋酸锡的组合,硫酸锡、氯化锡和醋酸锡的组合等。
作为本发明优选的技术方案,步骤(1)所述碱液包括尿素溶液、氢氧化钠溶液、碳酸钠溶液或碳酸钾溶液中的任意一种或至少两种的组合,所述组合典型但非限制性实例有:碳酸钠溶液与碳酸钾溶液的组合,碳酸钠溶液与尿素溶液的组合,碳酸钾溶液与氢氧化钠溶液的组合,尿素溶液、氢氧化钠溶液和碳酸钠溶液的组合,碳酸钠溶液、碳酸钾溶液、尿素溶液与氢氧化钠溶液的组合等。
优选地,步骤(1)所述碱液的加入方式为滴加。
优选地,步骤(1)所述碱液在搅拌条件下加入。
优选地,所述搅拌的速度为400~1200r/min,例如400r/min、600r/min、800r/min、1000r/min或1200r/min等,但并不仅限于所列举的数值,该数值范围内其他未列举的数值同样适用。
优选地,步骤(1)所述碱液的浓度为0.1~4mol/l,例如0.1mol/l、0.5mol/l、1.0mol/l、1.5mol/l、2.0mol/l、2.5mol/l、3.0mol/l、3.5mol/l或4.0mol/l等,但并不仅限于所列举的数值,该数值范围内其他未列举的数值同样适用。
优选地,步骤(1)所述碱液与所述混合溶液的体积比为1:3~4:1,例如1:3、1:2、3:4、1:1、3:2、2:1、3:1或4:1等,但并不仅限于所列举的数值,该数值范围内其他未列举的数值同样适用。
作为本发明优选的技术方案,步骤(2)所述老化的温度为-5~15℃,例如-5℃、-3℃、0℃、2℃、5℃、8℃、10℃、12℃或15℃等,但并不仅限于所列举的数值,该数值范围内其他未列举的数值同样适用。
本发明中,选择低温老化的原因在于:低温条件下,悬浊液中成核速率较慢,可以使成核颗粒大小均一,也可以使锡的分布更加均匀。
优选地,步骤(2)所述老化的时间为1~30h,例如1h、2h、4h、8h、12h、16h、20h、24h、28h或30h等,但并不仅限于所列举的数值,该数值范围内其他未列举的数值同样适用。
优选地,步骤(2)所述水热反应的温度为60~180℃,例如60℃、80℃、100℃、120℃、140℃、160℃或180℃等,但并不仅限于所列举的数值,该数值范围内其他未列举的数值同样适用。
优选地,步骤(2)所述水热反应的时间为0.5~24h,例如0.5h、1h、2h、4h、8h、12h、16h、20h或24h等,但并不仅限于所列举的数值,该数值范围内其他未列举的数值同样适用。
优选地,步骤(2)所述固液分离后,洗涤、干燥,得到第一固体产物。
本发明中,水热反应结束后一般冷却至15~30℃后,再进行分离。
优选地,所述干燥的温度为60~200℃,例如60℃、80℃、100℃、120℃、140℃、160℃、180℃或200℃等,但并不仅限于所列举的数值,该数值范围内其他未列举的数值同样适用。
优选地,所述干燥的时间为6~20h,例如6h、8h、10h、12h、14h、16h、18h或20h等,但并不仅限于所列举的数值,该数值范围内其他未列举的数值同样适用。
作为本发明优选的技术方案,步骤(3)所述分散液的溶剂为乙醇。
优选地,步骤(3)所述分散液中第一固体产物与乙醇的质量比为1:50~1:15,例如1:50、1:45、1:40、1:35、1:30、1:25、1:20或1:15等,但并不仅限于所列举的数值,该数值范围内其他未列举的数值同样适用。
作为本发明优选的技术方案,步骤(3)所述锌前驱体溶液中锌离子的浓度为0.26~1.60mmol/l,例如0.26mmol/l、0.40mmol/l、0.60mmol/l、0.80mmol/l、1.00mmol/l、1.20mmol/l、1.40mmol/l或1.60mmol/l等,但并不仅限于所列举的数值,该数值范围内其他未列举的数值同样适用。
优选地,步骤(3)所述锌前驱体为可溶性锌盐。
优选地,所述可溶性锌盐包括硝酸锌、硫酸锌、氯化锌或醋酸锌中的任意一种或至少两种的组合,所述组合典型但非限制性实例有:硝酸锌和氯化锌的组合,硝酸锌和醋酸锌的组合,硫酸锌和氯化锌的组合,硝酸锌、氯化锌和醋酸锌的组合,硫酸锌、氯化锌和醋酸锌的组合等。
优选地,步骤(3)所述锌前驱体溶液与分散液的体积比为(1~10):1,例如1:1、2:1、4:1、5:1、6:1、8:1或10:1等,但并不仅限于所列举的数值,该数值范围内其他未列举的数值同样适用。
优选地,步骤(3)所述锌前驱体溶液的加入方式为滴加。
优选地,步骤(3)所述锌前驱体溶液在搅拌条件下加入。
优选地,所述搅拌的速度为400~1200r/min,例如400r/min、600r/min、800r/min、1000r/min或1200r/min等,但并不仅限于所列举的数值,该数值范围内其他未列举的数值同样适用。
优选地,步骤(3)所述固液分离后,洗涤、干燥,得到第二固体产物。
优选地,所述干燥的温度为60~200℃,例如60℃、80℃、100℃、120℃、140℃、160℃、180℃或200℃等,但并不仅限于所列举的数值,该数值范围内其他未列举的数值同样适用。
优选地,所述干燥的时间为6~20h,例如6h、8h、10h、12h、14h、16h、18h或20h等,但并不仅限于所列举的数值,该数值范围内其他未列举的数值同样适用。
作为本发明优选的技术方案,步骤(4)所述焙烧的温度为300~1000℃,例如300℃、400℃、500℃、600℃、700℃、800℃、900℃或1000℃等,但并不仅限于所列举的数值,该数值范围内其他未列举的数值同样适用。
优选地,步骤(4)所述焙烧的时间为2~24h,例如2h、4h、6h、8h、10h、12h、16h、20h或24h等,但并不仅限于所列举的数值,该数值范围内其他未列举的数值同样适用。
优选地,步骤(4)所述焙烧在空气气氛和/或惰性气氛中进行。
本发明中,所述焙烧的目的在于分解锌前驱体,使之转化为锌单原子,焙烧气氛对产品的影响不大。
第三方面,本发明提供了一种上述双单原子助剂负载型氧化铜催化剂的用途,所述催化剂用于有机硅单体合成反应。
优选地,所述催化剂用于二甲基二氯硅烷的选择性合成。
二甲基二氯硅烷是制备有机硅材料最重要也是用量最大的单体,它是通过rochow反应,即一氯甲烷(mecl)和硅粉(si)在铜基催化剂的作用下直接反应得到,反应方程式为:
上述反应式中,m1为一甲基三氯硅烷,m2为二甲基二氯硅烷,m3为三甲基一氯硅烷,m1h为甲基氢二氯硅烷,m2h为二甲基氢一氯硅烷,lbr为低沸物,hbr为高沸物。
本发明所述催化剂的目的在于在提高该反应中m2产物的选择性和硅粉的转化率。
与现有技术相比,本发明具有以下有益效果:
(1)本发明所述双单原子助剂负载型氧化铜催化剂为首次合成,sn和zn单原子助剂分散性好,双单原子助剂间存在协同作用,与主催化剂cuo之间也存在较强的相互作用,稳定性强;
(2)本发明所述催化剂中sn和zn分别采用水热法和浸渍-焙烧法负载,反应条件容易控制,重复性好;
(3)本发明所述催化剂用于有机硅单体合成反应,与传统的纳米粒子助剂催化剂相比,表现出更加优异的催化性能,目标产物二甲基二氯硅烷选择性达到85.0%以上,硅粉原料转化率达到40.0%以上。
附图说明
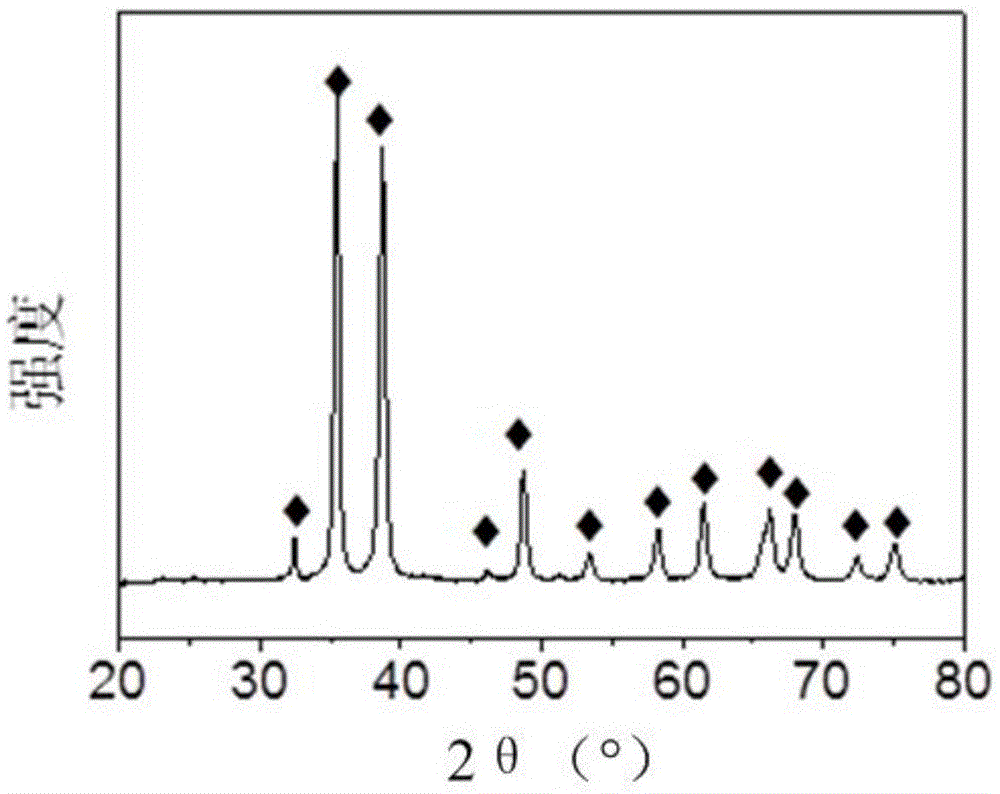
图1是本发明实施例1所述催化剂的xrd图;
图2是本发明实施例1所述催化剂的sem图;
图3是本发明实施例1所述催化剂的hrtem图;
图4是本发明实施例1所述催化剂的球差校正的原子分辨率的haadf-stem图;
图5是本发明实施例1所述催化剂的元素面分布图;
图6是本发明实施例1、对比例3和对比例5催化反应后废触体的xrd图,其中,内置图为矩形区域的放大图。
具体实施方式
为更好地说明本发明,便于理解本发明的技术方案,下面对本发明进一步详细说明。但下述的实施例仅是本发明的简易例子,并不代表或限制本发明的权利保护范围,本发明保护范围以权利要求书为准。
本发明具体实施方式部分提供了一种双单原子助剂负载型氧化铜催化剂及其制备方法,所述催化剂包括主催化剂cuo和单原子助剂sn和zn,所述单原子助剂负载在主催化剂表面上。
其制备方法包括如下步骤:
(1)向铜前驱体和锡前驱体混合溶液中加入碱液,得到悬浊液;
(2)将步骤(1)得到的悬浊液老化后进行水热反应,所得产物固液分离,得到第一固体产物;
(3)向步骤(2)得到的第一固体产物的分散液中加入锌前驱体溶液,固液分离得到第二固体产物;
(4)将步骤(3)得到的第二固体产物焙烧处理,得到双单原子助剂负载型氧化铜催化剂。
以下为本发明典型但非限制性实施例:
实施例1:
本实施例提供了一种双单原子助剂负载型氧化铜催化剂及其制备方法,所述催化剂包括主催化剂cuo和单原子助剂sn和zn,所述单原子助剂负载在主催化剂表面上。
所述催化剂的制备方法包括如下步骤:
(1)将10.70gcuso4·5h2o和0.01gsncl4·5h2o(n(cu):n(sn)=1500:1)溶于50ml水,搅拌得到澄清溶液,放入冰水浴中静置,直至溶液变为蓝色;
(2)向上述溶液中滴加100ml0.1mol/l的氢氧化钠溶液,在1000r/min转速下搅拌0.5h,得到悬浊液;
(3)将步骤(2)得到的悬浊液在0℃条件下老化4h后,转移至200ml的聚四氟乙烯内衬的高压釜中,于130℃下水热反应18h,冷却后过滤,所得固体用去离子水和无水乙醇洗涤数次,之后于60℃下干燥12h,得到第一固体产物;
(4)取1g第一固体产物分散于乙醇中,逐滴加入20ml浓度为0.26mmol/l的氯化锌水溶液,在1000r/min转速下搅拌0.5h,过滤分离,所得固体用去离子水和无水乙醇洗涤数次,之后于60℃下干燥12h,得到第二固体产物;
(5)将步骤(4)得到的第二固体产物在空气气氛下400℃焙烧18h,得到双单原子助剂负载型氧化铜催化剂。
将制得的催化剂采用荷兰panalytical公司(帕纳科)生产的x′pertprompd型多功能x射线衍射仪进行xrd测试,结果如图1所示;将制得的催化剂采用日本jeol公司生产的jsm-7001f型扫描电子显微镜观察其微观形貌,其sem图如图2所示;将制得的催化剂采用日本jeol公司生产的jem-2010f型透射电子显微镜上观察其内部结构,其hrtem图如图3所示;将制得的催化剂采用扫描透射电子显微镜进行测试,其球差校正的原子分辨率的haadf-stem图如图4所示;将制得的催化剂采用英国牛津公司生产的incax-max型能谱仪测试元素分布情况,其元素面分布图如图5所示;将制得的催化剂采用美国pekin-elmer电感耦合等离子体原子发射光谱仪进行icp测试。
本实施例中,由图1可知,所述催化剂的衍射峰均与“◆”相对应,而“◆”为cuo的特征衍射峰,表明所述催化剂只有cuo的衍射峰,无zn和sn对应的衍射峰出现;由图2可知,所述催化剂呈片状,径向尺寸约为1μm,厚度约为40nm;从图3中可以看到清晰的晶格条纹,晶面间距值对应cuo的(110)晶面,没有观察到zn和sn对应的晶格条纹,表明zn和sn高度分散在cuo中;图4可进一步证明图3的结果,同时由于元素衬度的差别,还可以观察到许多小的亮点;由图5可知,催化剂中zn和sn在催化剂表面均匀分布;由icp测试结果可知,催化剂中铜元素含量为79.2wt%,锡元素含量为0.15wt%,锌元素含量为0.20wt%。上述分析表明,zn和sn以单原子的形式分散存在。
实施例2:
本实施例提供了一种双单原子助剂负载型氧化铜催化剂及其制备方法,所述催化剂包括主催化剂cuo和单原子助剂sn和zn,所述单原子助剂负载在主催化剂表面上。
所述催化剂的制备方法包括如下步骤:
(1)将12.84gcuso4·5h2o和0.01gsncl4·5h2o(n(cu):n(sn)=1800:1)溶于50ml水,搅拌得到澄清溶液,放入冰水浴中静置,直至溶液变为蓝色;
(2)向上述溶液中滴加100ml0.5mol/l的尿素溶液,在500r/min转速下搅拌1h,得到悬浊液;
(3)将步骤(2)得到的悬浊液在-5℃条件下老化20h后,转移至200ml的聚四氟乙烯内衬的高压釜中,于60℃下水热反应24h,冷却后过滤,所得固体用去离子水和无水乙醇洗涤数次,之后于100℃下干燥8h,得到第一固体产物;
(4)取1g第一固体产物分散于乙醇中,逐滴加入20ml浓度为0.52mmol/l的氯化锌水溶液,在500r/min转速下搅拌1h,过滤分离,所得固体用去离子水和无水乙醇洗涤数次,之后于100℃下干燥8h,得到第二固体产物;
(5)将步骤(4)得到的第二固体产物在氮气气氛下600℃焙烧12h,得到双单原子助剂负载型氧化铜催化剂。
将制得的催化剂采用电感耦合等离子体原子发射光谱仪进行icp测试,由icp测试结果可知,催化剂中铜元素含量为79.8wt%,锡元素含量为0.04wt%,锌元素含量为0.05wt%。
实施例3:
本实施例提供了一种双单原子助剂负载型氧化铜催化剂及其制备方法,所述催化剂包括主催化剂cuo和单原子助剂sn和zn,所述单原子助剂负载在主催化剂表面上。
所述催化剂的制备方法包括如下步骤:
(1)将14.97gcuso4·5h2o和0.01gsncl4·5h2o(n(cu):n(sn)=2100:1)溶于100ml水,搅拌得到澄清溶液,放入冰水浴中静置,直至溶液变为蓝色;
(2)向上述溶液中滴加50ml2mol/l的碳酸钠溶液,在800r/min转速下搅拌0.5h,得到悬浊液;
(3)将步骤(2)得到的悬浊液在15℃条件下老化1h后,转移至200ml的聚四氟乙烯内衬的高压釜中,于180℃下水热反应4h,冷却后过滤,所得固体用去离子水和无水乙醇洗涤数次,之后于200℃下干燥6h,得到第一固体产物;
(4)取1g第一固体产物分散于乙醇中,逐滴加入20ml浓度为0.78mmol/l的硝酸锌水溶液,在800r/min转速下搅拌0.5h,过滤分离,所得固体用去离子水和无水乙醇洗涤数次,之后于200℃下干燥6h,得到第二固体产物;
(5)将步骤(4)得到的第二固体产物在氩气气氛下1000℃焙烧2h,得到双单原子助剂负载型氧化铜催化剂。
将制得的催化剂采用电感耦合等离子体原子发射光谱仪进行icp测试,由icp测试结果可知,催化剂中铜元素含量为79.98wt%,锡元素含量为0.016wt%,锌元素含量为0.1wt%。
实施例4:
本实施例提供了一种双单原子助剂负载型氧化铜催化剂及其制备方法,所述催化剂包括主催化剂cuo和单原子助剂sn和zn,所述单原子助剂负载在主催化剂表面上。
所述催化剂的制备方法包括如下步骤:
(1)将10.91gcu(no3)2·3h2o和0.01gsn(ch3coo)4(n(cu):n(sn)=1600:1)溶于50ml水,搅拌得到澄清溶液,放入冰水浴中静置,直至溶液变为蓝色;
(2)向上述溶液中滴加50ml4mol/l的尿素溶液,在1000r/min转速下搅拌0.5h,得到悬浊液;
(3)将步骤(2)得到的悬浊液在5℃条件下老化8h后,转移至150ml的聚四氟乙烯内衬的高压釜中,于150℃下水热反应8h,冷却后过滤,所得固体用去离子水和无水乙醇洗涤数次,之后于80℃下干燥10h,得到第一固体产物;
(4)取1g第一固体产物分散于乙醇中,逐滴加入15ml浓度为1.4mmol/l的硫酸锌水溶液,在1000r/min转速下搅拌0.5h,过滤分离,所得固体用去离子水和无水乙醇洗涤数次,之后于80℃下干燥10h,得到第二固体产物;
(5)将步骤(4)得到的第二固体产物在氩气气氛下300℃焙烧24h,得到双单原子助剂负载型氧化铜催化剂。
将制得的催化剂采用电感耦合等离子体原子发射光谱仪进行icp测试,由icp测试结果可知,催化剂中铜元素含量为79.3wt%,锡元素含量为0.75wt%,锌元素含量为0.2wt%。
实施例5:
本实施例提供了一种双单原子助剂负载型氧化铜催化剂及其制备方法,所述催化剂包括主催化剂cuo和单原子助剂sn和zn,所述单原子助剂负载在主催化剂表面上。
所述催化剂的制备方法包括如下步骤:
(1)将8.29gcucl2·2h2o和0.01gsncl4·5h2o(n(cu):n(sn)=1700:1)溶于50ml水,搅拌得到澄清溶液,放入冰水浴中静置,直至溶液变为蓝色;
(2)向上述溶液中滴加200ml0.1mol/l的氢氧化钠溶液,在1000r/min转速下搅拌0.5h,得到悬浊液;
(3)将步骤(2)得到的悬浊液在10℃条件下老化3h后,转移至300ml的聚四氟乙烯内衬的高压釜中,于100℃下水热反应12h,冷却后过滤,所得固体用去离子水和无水乙醇洗涤数次,之后于80℃下干燥10h,得到第一固体产物;
(4)取1g第一固体产物分散于乙醇中,逐滴加入20ml浓度为1.6mmol/l的硫酸锌水溶液,在1000r/min转速下搅拌0.5h,过滤分离,所得固体用去离子水和无水乙醇洗涤数次,之后于80℃下干燥10h,得到第二固体产物;
(5)将步骤(4)得到的第二固体产物在氖气气氛下800℃焙烧6h,得到双单原子助剂负载型氧化铜催化剂。
将制得的催化剂采用电感耦合等离子体原子发射光谱仪进行icp测试,由icp测试结果可知,催化剂中铜元素含量为79.5wt%,锡元素含量为0.45wt%,锌元素含量为0.4wt%。
实施例6:
本实施例提供了一种双单原子助剂负载型氧化铜催化剂及其制备方法,所述催化剂包括主催化剂cuo和单原子助剂sn和zn,所述单原子助剂负载在主催化剂表面上。
所述催化剂的制备方法包括如下步骤:
(1)将12.86gcu(ch3coo)2·h2o和0.01gsn(so4)2(n(cu):n(sn)=2000:1)溶于50ml水,搅拌得到澄清溶液,放入冰水浴中静置,直至溶液变为蓝色;
(2)向上述溶液中滴加100ml1mol/l的碳酸钾溶液,在1000r/min转速下搅拌0.5h,得到悬浊液;
(3)将步骤(2)得到的悬浊液在2℃条件下老化4h后,转移至200ml的聚四氟乙烯内衬的高压釜中,于80℃下水热反应20h,冷却后过滤,所得固体用去离子水和无水乙醇洗涤数次,之后于80℃下干燥10h,得到第一固体产物;
(4)取1g第一固体产物分散于乙醇中,逐滴加入20ml浓度为1.0mmol/l的醋酸锌水溶液,在1000r/min转速下搅拌0.5h,过滤分离,所得固体用去离子水和无水乙醇洗涤数次,之后于80℃下干燥10h,得到第二固体产物;
(5)将步骤(4)得到的第二固体产物在氩气气氛下500℃焙烧14h,得到双单原子助剂负载型氧化铜催化剂。
将制得的催化剂采用电感耦合等离子体原子发射光谱仪进行icp测试,由icp测试结果可知,催化剂中铜元素含量为79.7wt%,锡元素含量为0.1wt%,锌元素含量为0.8wt%。
对比例1:
本对比例提供了一种氧化铜催化剂的制备方法,所述催化剂的制备方法参照实施例1,区别仅在于:步骤(1)中不加入锡前驱体sncl4·5h2o,也不包括步骤(4)和(5)。
对比例2:
本对比例提供了一种单原子sn助剂负载的氧化铜催化剂的制备方法,所述催化剂的制备方法参照实施例1,区别仅在于:不包括步骤(4)和(5)。
将制得的催化剂采用电感耦合等离子体原子发射光谱仪进行icp测试,由icp测试结果可知,催化剂中铜元素含量为79.7wt%,锡元素含量为0.1wt%。
对比例3:
本对比例提供了一种氧化铜基催化剂,所述催化剂由氧化铜和氧化锡机械混合组成,其中,所述氧化铜采用对比例1中的方法制备得到,所述氧化锡为商业氧化锡,两者混合的质量比为20:1。
对比例4:
本对比例提供了一种单原子sn助剂和zn纳米粒子负载的氧化铜催化剂的制备方法,所述催化剂的制备方法参照实施例1,区别仅在于:步骤(4)中所加氯化锌水溶液的浓度为2.0mmol/l。
将制得的催化剂进行icp测试,由icp测试结果可知,催化剂中铜元素含量为79.7wt%,锡元素含量为0.1wt%,锌元素含量为1.5wt%。
对比例5:
本对比例提供了一种商业氧化铜催化剂。
将实施例1-6和对比例1-5所得催化剂用于催化一氯甲烷和硅粉反应生成二甲基二氯硅烷的反应,来评价催化剂的催化性能。催化剂性能评价实验采用微型固定床装置进行,反应器内径为20cm,长度为50cm,评价过程如下:将10gsi粉和0.5g制得的催化剂研磨混合形成触体;反应时,首先采用n2吹扫反应系统,然后,切换为mecl气体,经过预热后与触体发生反应,反应条件为:预热温度为350℃,反应温度为325℃,反应压力为常压,mecl的流速为25ml/min,反应时间为24h。
反应后的产物经冷凝管冷凝后采用甲苯收集,多余尾气用碱液吸收;收集的混合液定容后采用气相色谱(agilent7890b,kb-210色谱柱,tcd检测器)进行定量分析。
上述实施例和对比例中的催化剂的活性测试结果如表1所示,其中,产物分布通过气相色谱分析结果中反应产物对应面积的百分比计算,硅转化率的计算公式为:
其中,w为触体的重量。
将实施例1、对比例3和对比例5催化反应后形成的废触体进行xrd测试,其xrd图如图6所示,其中内置图为图中矩形区域的放大图。
表1催化剂活性测试结果表
由表1可以看出,实施例1-6中制备的催化剂的催化活性较高,m2的选择性均达到85.0%以上,si粉转化率均达到40.0%以上,其中m2选择性最优值达到89.5%,si粉转化率最优值为48.2%;而对比例1中制备的单一氧化铜催化剂的m2选择性仅为31.2%,si粉转化率仅为3.2%;对比例2中负载单原子助剂sn后,m2选择性明显上升,si粉转化率虽也提高,但与实施例仍有差距;对比例3中采用氧化铜和氧化锡直接混合的方式,虽然sn含量明显高于对比例2,但其m2的选择性与对比例2相比降低,此时sn未以单原子助剂的形式存在,与氧化铜之间的作用较弱;对比例4中,zn含量偏高,以纳米粒子形式负载,无法充分利用单原子的优势,低于实施例中的催化活性;对比例5中商业氧化铜催化剂的m2选择性为75.0%,si粉转化率仅为28.5%,也明显弱于本发明实施例。除此之外,单独使用zn作为助剂时,由于其与cu之间的作用,易形成异质结构,而难以形成本发明中的单原子助剂催化剂。
以上实施例和对比例结果表明,本发明提供的催化剂在催化性能方面具有显著优势,其主要原因在于:本发明所述催化剂中助剂以单原子形式存在,与纳米粒子相比,与主催化剂之间具有更强的相互作用;同时所述催化剂的双单原子助剂之间具有协同催化的作用,sn和cu共同作为ch3的吸附位,加强了氯甲烷的吸附,zn对cu的电子结构产生影响,使其更偏向于低价态cu,可以进一步促进氯甲烷在cu位上的吸附,以上特征使得活性铜原子快速形成,以产生更多的cu3si活性相,如图6所示,实施例1中所述催化剂含有的cu3si的衍射峰,提高m2选择性和硅粉转化率;而对比例3中cu3si的衍射峰强度较弱,还含有cu6.69si的衍射峰,是其催化活性相对减弱。
申请人声明,本发明通过上述实施例来说明本发明的制备方法和应用,但本发明并不局限于上述方法和应用,即不意味着本发明必须依赖上述方法和应用才能实施。所属技术领域的技术人员应该明了,对本发明的任何改进,对本发明所用原料、操作的等效替换及辅助成分的添加、具体方式的选择等,均落在本发明的保护范围和公开范围之内。
- 还没有人留言评论。精彩留言会获得点赞!