一种用于取代酮和双酚F合成的负载型钯催化剂的制作方法
一种用于取代酮和双酚f合成的负载型钯催化剂
技术领域
[0001]
本发明涉及一种用于取代酮和双酚f合成的负载型钯催化剂,属于化学材料与药物领域。
背景技术:
[0002]
α,β-不饱和羰基化合物及其衍生物是一类具有生物活性的化合物,其结构广泛存在于药物分子、功能材料和天然产物中,常常用于合成杂环和芳香化合物,更是各种手性化合物的前手性分子。
[0003]
绿色合成具有经济性、环境友好和高效性等特点,是目前研究的热点领域。α,β-不饱和羰基化合物是一类重要的有机合成前体,其中aza-michael和michael加成反应分别是构建c-n键和c-c键的经典有机合成方法,但传统方法的苛刻条件难以满足目前绿色化学的要求,亟待发展新的绿色合成方法,高效地构建目标化合物,从而减小对环境的污染。此外,利用过渡金属催化的反应来合成α,β-不饱和羰基化合物的方法近些年也有诸多报导,但通常需要在强酸或者钯属催化剂及加热的条件下进行,传统催化剂往往存在着活性低、选择性差、对环境造成污染等缺点,所以急需发展温和的催化体系合成α,β-不饱和羰基化合物。随着金属有机化学的发展,出现了很多可以作为均相催化剂的金属络合物,但此类催化剂对反应器有腐蚀作用,在空气和水中稳定性差,催化剂的分离和回收困难,其应用受到一定的限制。随着材料领域的发展,负载型催化剂的研究更加深入,负载型催化剂基本兼具无机物非均相催化剂和金属有机均相催化剂的优点,它不但具有较高的活性和选择性,腐蚀性小,而且容易回收重复利用,且稳定性好。
技术实现要素:
[0004]
为了解决现有技术存在的问题,本发明提供了一种用于炔丙醇重排合成取代酮和双酚f类衍生物的负载型钯催化剂及其制备方法,本发明首先以(1h-苯并三唑-1-基)乙酸配体,与钯合成得到了钯催化剂triapdcl,最后将triapdcl负载在石墨烯上,得到了负载型钯催化剂triapdcl@go。本发明的催化剂在温和条件下对炔丙醇类化合物的重排反应具有优异的催化活性和选择性,产率都在80%以上,产物的e式产物的选择性在40:1以上。符合绿色化学的要求,克服了均相催化剂难以分离和回收的缺点,能够回收重复利用,此外,本发明的催化剂还能够用于合成双酚f的反应,双酚f产物的选择性在95%以上。用途多样且催化性能优异,具有良好的应用场景。
[0005]
本发明的第一个目的是提供一种制备用于取代酮和双酚f合成的的负载型钯催化剂triapdcl@go的方法,所述方法包括如下步骤:
[0006]
(1)将(1h-苯并三唑-1-基)乙酸、醋酸钠和钯源分散在溶剂中,在20-80℃下进行反应,反应结束后,冷却,固液分离、收集清液,浓缩干燥,制得钯催化剂前体(triapdx);
[0007]
(2)将钯催化剂前体(triapdx)、石墨烯分散于有机溶剂中,在140-160℃下进行反应,反应结束后,冷却,固液分离、收集固体,即得负载型钯催化剂(triapdx@go)。
[0008]
在本发明的一种实施方式中,所述步骤(1)中的溶剂包括乙醇、甲醇、四氢呋喃、乙腈中任意一种或多种。
[0009]
在本发明的一种实施方式中,所述步骤(1)中的钯源为pdx2;x为cl、br、i、oac。
[0010]
在本发明的一种实施方式中,所述步骤(1)中(1h-苯并三唑-1-基)乙酸相对溶剂的浓度为0.1~1mmol/ml。
[0011]
在本发明的一种实施方式中,所述步骤(1)中(1h-苯并三唑-1-基)乙酸与醋酸钠的摩尔比为1:1。
[0012]
在本发明的一种实施方式中,所述步骤(1)中(1h-苯并三唑-1-基)乙酸与钯源摩尔比的用量比为1:1。
[0013]
在本发明的一种实施方式中,步骤(1)中所述的反应是在120-480r/min转速下进行的。
[0014]
在本发明的一种实施方式中,所述步骤(1)中还包括:固液分离、收集固体后,将固体在溶解在二氯甲烷中,并用石油醇或者正己烷进行重结晶,固液分离、收集固体,干燥。
[0015]
在本发明的一种实施方式中,所述步骤(2)中钯催化剂前体与石墨烯的质量比为1:50。
[0016]
在本发明的一种实施方式中,所述步骤(2)中钯催化剂前体相对有机溶剂的浓度为(5-10)mg/ml。
[0017]
在本发明的一种实施方式中,所述步骤(2)中有机溶剂包括dmso、dmf、dma中任意一种或多种。
[0018]
在本发明的一种实施方式中,步骤(2)中所述的反应是200-800r/min转速下进行的。
[0019]
在本发明的一种实施方式中,所述步骤(2)中还包括:对收集的固体进行洗涤、干燥。
[0020]
在本发明的一种实施方式中,所述步骤制备方法具体包括如下步骤:
[0021]
(1)将2-(1h-苯并三唑-1-基)乙酸和乙醇混合均匀,然后再加入与(1h-苯并三唑-1-基)乙酸摩尔比为1:1的醋酸钠,搅拌反应30-60min;
[0022]
(2)向步骤(1)得到的反应溶液中加入pdcl,在20-80℃下搅拌反应12-24h,反应结束后冷却、过滤,将过滤得到的上清液通过真空浓缩得到粗产品;
[0023]
(3)将步骤(2)所得的粗产品溶解在二氯甲烷中,并用石油醇或者正己烷进行重结晶,固液分离、干燥即可得到钯催化剂前体triapdx;
[0024]
(4)在dmso中加入步骤(3)所得的催化剂triapdcl和石墨烯,在140-160℃下搅拌反应12-16h,反应结束后冷却,离心,用蒸馏水和无水乙醇分别洗涤2-3次,真空干燥,得到负载型钯催化剂triapdx@go。
[0025]
在本发明的一种实施方式中,步骤(2)中所述过滤优选为通过硅藻土进行过滤。
[0026]
在本发明的一种实施方式中,步骤(3)中所述固液分离为过滤或离心分离。
[0027]
在本发明的一种实施方式中,步骤(3)(4)中所述干燥优选为真空干燥,优选温度为30℃,时间为24h。
[0028]
本发明的第二个目的是利用上述方法制备得到一种负载型钯催化剂(triapdx@go)。
[0029]
本发明的第三个目的是提供上述的负载型钯催化剂应用在催化炔丙醇类化合物重排、催化合成双酚f中。
[0030]
本发明的第四个目的是提供一种制备取代酮类化合物的方法,所述方法为:上述的负载型钯催化剂triapdx@go作为催化剂。
[0031]
在本发明的一种实施方式中,所述方法具体为:将炔丙醇类化合物、上述制备得到的钯催化剂triapdx@go和醇类化合物混合,在40-60℃下反应6-24小时,冷却、除去溶剂、纯化即可得到相应的取代酮类化合物。
[0032]
在本发明的一种实施方式中,所述炔丙醇类化合物的结构如下所示:
[0033][0034]
其中,r1、r2分别独立地选自:氢、c1-8烷基、烷氧基、卤素、卤代烷基、芳基。(芳基中的取代基包括氢、c1-8烷基、烷氧基、卤素、卤代烷基;芳基包括苯环或者萘环)
[0035]
在本发明的一种实施方式中,所述炔丙醇类化合物的结构优选如下结构:
[0036][0037]
其中,r1’
、r2’
分别独立地选自:氢、c1-8烷基、烷氧基、卤素、卤代烷基。
[0038]
在本发明的一种实施方式中,所述醇类化合物的结构为:r
3-oh;r3选自c1-8烷基、芳基取代的c1-8烷、芳基。(芳基中的取代基包括氢、c1-8烷基、烷氧基、卤素、卤代烷基;芳基包括苯环或者萘环)
[0039]
在本发明的一种实施方式中,所述取代酮类化合物制备的反应路线为:
[0040][0041]
在本发明的一种实施方式中,所述钯催化剂triapdx@go的用量为炔丙醇类化合物的0.01-0.1倍摩尔当量。
[0042]
在本发明的一种实施方式中,所述醇类化合物为甲醇或乙醇,优选乙醇。
[0043]
在本发明的一种实施方式中,所述反应温度为40~60℃。具有可选择40、50、60℃。
[0044]
在本发明的一种实施方式中,反应后优选利用tlc(薄层色谱法)检测反应是否结束。
[0045]
本发明的第五个目的是提供一种合成双酚f类化合物的方法,所述方法以上述的钯催化剂triapdx@go为催化剂。
[0046]
在本发明的一种实施方式中,所述合成双酚f的反应路线为:
[0047][0048]
在本发明的一种实施方式中,所述方法为在反应容器中加入苯酚、上述制备得到
的钯催化剂triapdx@go和甲醛,在35-110℃下反应1-36h,用碳酸氢钠调ph为5-6,分离有机相,随后溶解于naoh溶液中,用浓盐酸调ph约为3-6,即得到产品双酚f化合物。
[0049]
在本发明的一种实施方式中,所述苯酚、钯催化剂、甲醛的摩尔比为10:3:5。
[0050]
与现有技术相比,本发明具有以下优势:
[0051]
(1)本发明的催化剂无需用到大量的酸,大大降低了成本,尤其是避免了废酸的产生,减少环境污染,可工业化生产。
[0052]
(2)本发明制备得到的钯催化剂在温和条件下即进行炔丙醇类化合物的重排反应,具有优异的催化活性,对应的α,β-不饱和羰基化合物的收率达80%以上。本发明制备的钯催化剂克服了其他贵金属催化剂难以分离,不能重复使用的特点,反应过程更加绿色环保,符合绿色化学的要求。
[0053]
(3)本发明制备得到的钯催化剂还能够用于合成双酚f的反应,用途多样且催化性能优异,具有更广泛的应用场景。
附图说明
[0054]
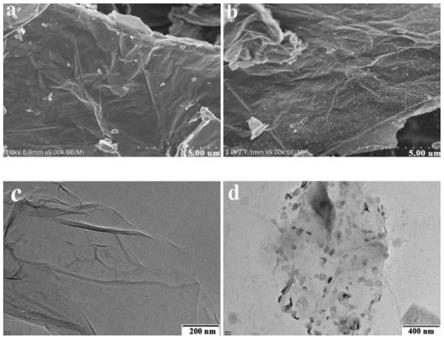
图1为实施例1所得负载型钯催化剂的电镜图;其中,a、b均为负载型钯催化剂的扫描电镜图;c、d均为负载型钯催化剂的透射电镜图。
具体实施方式
[0055]
收率的计算公式:收率=(实际产物质量/理论产物质量)
×
100%;
[0056]
以下,申请人对本发明做了具体实验,并且详细描述了这种钯催化剂应用在炔丙醇类化合物重排反应的实验过程。这些仅用于详尽说明本发明,并不以任何方式限制发明的范围。
[0057]
本发明涉及的配体(1h-苯并三唑-1-基)乙酸的来源:参照design and synthesis of alanine triazole ligands and application in promotion of hydration,allene synthesis and borrowing hydrogen reactions(advanced synthesis&catalysis,358(9),1433-1439;2016)中方法,在thf/meoh(1:1)和lioh水溶液的混合溶液中添加1h-苯并三唑-1-羧酸甲酯,在室温下搅拌1-2小时后,加入hcl水溶液,酸化至ph至2~3,旋蒸除去溶剂,抽滤得到(1h-苯并三唑-1-基)乙酸。
[0058]
实施例1负载型钯催化剂的制备
[0059]
(1)将(1h-苯并三唑-1-基)乙酸(1.0mmol),etoh(10ml)加入圆底烧瓶中,搅拌均匀,然后再加入naoac(1.0mmol),室温下搅拌反应45min。然后向上述溶液中加入pdcl2(1.0mmol),反应混合物在25℃下搅拌反应12h,待反应结束冷却至室温。反应结束后,将浑浊液通过硅藻土进行过滤,澄清溶液通过真空减压除去溶剂得到粗产品。将所得粗产品进一步溶解在dcm(10ml)中,并用石油醇(50ml)进行重结晶,再次抽滤,真空干燥得到triapdcl。
[0060]
(2)将步骤(1)制得的triapdcl(1.0mmol),石墨烯(1.0g),dmso(30ml)加入圆底烧瓶中,160℃下搅拌反应12h,待反应结束后冷却至室温,将浑浊液进行离心,除去上清液,用蒸馏水和无水乙醇洗涤3次,真空干燥,得到负载型钯催化剂triapdcl@go。
[0061]
实施例2负载型钯催化剂的制备
[0062]
(1)将(1h-苯并三唑-1-基)乙酸(1.0mmol),etoh(10ml)加入圆底烧瓶中,搅拌均匀,然后再加入naoac(1.0mmol),室温下搅拌反应60min。然后向上述溶液中加入pdcl2(1.0mmol),反应混合物在60℃下搅拌反应12h,待反应结束冷却至室温。反应结束后,将浑浊液通过硅藻土进行过滤,澄清溶液通过真空减压除去溶剂得到粗产品。将所得粗产品进一步溶解在dcm(10ml)中,并用正己烷(50ml)进行重结晶,再次抽滤,真空干燥得到triapdcl。
[0063]
(2)将步骤(1)制得的triapdcl(1.0mmol),石墨烯(1.0g),dmso(30ml)加入圆底烧瓶中,160℃下搅拌反应12h,待反应结束后冷却至室温,将浑浊液进行离心,除去上清液,用蒸馏水和无水乙醇洗涤3次,真空干燥,得到负载型钯催化剂triapdcl@go。
[0064]
实施例3苯基丙-2-烯-1-酮的制备
[0065]
将1,3-二苯基-2-丙炔-1-醇(0.5mol)、实施例1制备得到的triapdcl@go催化剂(0.05mol)、2ml甲醇加入到反应容器中,反应混合物在60℃下搅拌反应6小时后,冷却至室温,减压除去溶剂,用硅胶柱层析(石油醚/乙酸乙酯)对粗品进行纯化,得到产物苯基丙-2-烯-1-酮,收率91%。
[0066]
产物的选择性e/z=41:1。
[0067]
苯基丙-2-烯-1-酮的表征数据:
[0068]1h nmr(400mhz,cdcl3):δ7.57
–
7.53(m,3h),7.45
–
7.33(m,3h),6.75(d,j=16.0hz,1h),2.67(t,j=7.0hz,2h),1.71
–
1.60(m,2h),1.33(m,2h),0.93(t,j=7.0hz,3h);
13
c nmr(101mhz,cdcl3):δ200.6,142.2,134.6,130.34,128.7,128.3,126.3,40.7,26.4,22.3,13.8.
[0069]
实施例4三苯基丙-2-烯-1-酮的制备
[0070]
将三苯基丙-2-炔-1-醇(0.5mol)、实施例1制备得到的triapdcl@go催化剂(0.05mol)、2ml乙醇加入到反应容器中,反应混合物在80℃下搅拌反应4小时后,冷却至室温,减压除去溶剂,用硅胶柱层析(石油醚/乙酸乙酯)对粗品进行纯化,得到产物三苯基丙-2-烯-1-酮,收率82%。
[0071]
产物的选择性e/z=54:1。
[0072]
三苯基丙-2-烯-1-酮的表征数据:
[0073]1h nmr(600mhz,cdcl3)δ7.97(d,j=7.8hz,2h),7.51(t,j=7.2hz,1h),7.47
–
7.38(m,7h),7.33
–
7.29(m,3h),7.26
–
7.22(m,2h),7.17(s,1h);
[0074]
13
c nmr(151mhz,cdcl3):δ192.3,154.4,141.0,138.7,137.8,132.3,129.4,129.0,128.4,128.2,128.1,128.0,127.7,123.7.
[0075]
实施例5 1-(2-氯苯基)庚-1-烯-3-酮的制备
[0076]
将1-氯-2-(辛基-3-炔-2-苯基)-2-醇(0.5mol)、实施例1制备得到的triapdcl@go催化剂(0.05mol)、2ml甲醇加入到反应容器中,反应混合物在60℃下搅拌反应6小时后,冷却至室温,减压除去溶剂,用硅胶柱层析(石油醚/乙酸乙酯)对粗品进行纯化,得到产物1-(2-氯苯基)庚-1-烯-3-酮,收率82%。
[0077]
产物的选择性e/z=42:1。
[0078]
1-(2-氯苯基)庚-1-烯-3-酮的表征数据:
[0079]1h nmr(400mhz,cdcl3)δ7.97(d,j=16.0hz,1h),7.65(dd,j=7.2,2.0hz,1h),
7.44(dd,j=8.0,2.0hz,1h),7.38
–
7.27(m,2h),6.71(d,j=16.0hz,1h),2.72(t,j=7.2hz,2h),1.74
–
1.62(m,2h),1.47
–
1.35(m,2h),0.97(t,j=7.2hz,3h);
13
c nmr(101mhz,cdcl3):δ200.5,138.1,135.2,132.9,131.1,130.2,128.9,127.6,127.1,40.2,26.5,22.4,13.9.
[0080]
实施例6丁基苯乙烯基酮的制备
[0081]
将1-苯基-2-庚炔-1-醇(0.5mol),实施例1制备得到的triapdcl@go催化剂(0.05mol),3ml乙醇加入到反应容器中,反应混合物在60℃下搅拌反应12小时后,冷却至室温,减压除去溶剂,用硅胶柱层析(石油醚/乙酸乙酯)对粗品进行纯化,得到产物丁基苯乙烯基酮,收率89%。
[0082]
产物的选择性e/z=58:1。
[0083]
丁基苯乙烯基的表征数据:
[0084]1h nmr(400mhz,cdcl3):δ7.58
–
7.51(m,3h),7.43
–
7.36(m,3h),6.74(d,j=16.2hz,1h),2.66(t,j=7.2hz,2h),1.70
–
1.62(m,2h),1.37(m,2h),0.94(t,j=7.2hz,3h);
13
c nmr(101mhz,cdcl3):δ200.7,142.3,134.5,130.3,128.9,128.2,126.2,40.6,26.5,22.4,13.9.
[0085]
由此实施例可见,当乙醇参与反应时,同样能够得到较高的收率。故该负载型钯催化剂的参与下,能够将乙醇替代有毒的甲醇参与反应,更加绿色环保。
[0086]
实施例7催化合成双酚f
[0087]
在50ml烧瓶中加入5mmol苯酚,加热至完全融化,加入实施例3制备得到的triapdcl@go催化剂1.0mmol),再滴加入3mmol的甲醛溶液,在40℃下反应6h,加入适量碳酸氢钠调ph约为5-6,分离有机相,减压蒸馏,将粗产品溶解于适量naoh溶液中,用浓盐酸调ph约为6,即得到产品双酚f,产率85%,双酚f的选择性>95%。
[0088]
双酚f的表征数据:
[0089]1h nmr(400mhz,acetone-d6,δppm):3.81(s,2h),6.73(m,4h),7.04(m,4h),8.11(s,2h).
[0090]
对比例1
[0091]
探究不同催化剂对催化合成取代酮类化合物的影响:
[0092]
催化剂a:参照实施例1,将步骤(1)中配体由(1h-苯并三唑-1-基)乙酸替换为1,1'-双(二苯基膦)二茂铁,其他条件不变:
[0093]
将1,1'-双(二苯基膦)二茂铁(1.0mmol),etoh(10ml)加入圆底烧瓶中,搅拌均匀,然后再加入naoac(1.0mmol),室温下搅拌反应45min。然后向上述溶液中加入pdcl2(1.0mmol),反应混合物在25℃下搅拌反应12h,待反应结束冷却至室温。反应结束后,将浑浊液通过硅藻土进行过滤,澄清溶液通过真空减压除去溶剂得到粗产品。将所得粗产品进一步溶解在dcm(10ml)中,并用石油醇(50ml)进行重结晶,再次抽滤,真空干燥得到配体-pdcl2;
[0094]
将步骤(1)制得的配体-pdcl2(1.0mmol),石墨烯(1.0g),dmso(30ml)加入圆底烧瓶中,160℃下搅拌反应12h,待反应结束后冷却至室温,将浑浊液进行离心,除去上清液,用蒸馏水和无水乙醇洗涤3次,真空干燥,得到负载型钯催化剂a,备用。
[0095]
催化剂b:参照实施例1,将步骤(1)中pdcl2替换为cucl2,其他条件不变:
[0096]
(1)将(1h-苯并三唑-1-基)乙酸(1.0mmol),etoh(10ml)加入圆底烧瓶中,搅拌均匀,然后再加入naoac(1.0mmol),室温下搅拌反应45min。然后向上述溶液中加入cucl2(1.0mmol),反应混合物在25℃下搅拌反应12h,待反应结束冷却至室温。反应结束后,将浑浊液通过硅藻土进行过滤,澄清溶液通过真空减压除去溶剂得到粗产品。将所得粗产品进一步溶解在dcm(10ml)中,并用石油醇(50ml)进行重结晶,再次抽滤,真空干燥得到triacucl2。
[0097]
(2)将步骤(1)制得的triacucl2(1.0mmol),石墨烯(1.0g),dmso(30ml)加入圆底烧瓶中,160℃下搅拌反应12h,待反应结束后冷却至室温,将浑浊液进行离心,除去上清液,用蒸馏水和无水乙醇洗涤3次,真空干燥,得到负载型钯催化剂triacucl@go,备用。
[0098]
参照实施例3的过程,对上述得到的催化剂a、b的催化性能进行探究:
[0099]
将1,3-二苯基-2-丙炔-1-醇(0.5mol)、催化剂(0.05mol)、2ml甲醇加入到反应容器中,反应混合物在60℃下搅拌反应6小时后,冷却至室温,减压除去溶剂,用硅胶柱层析(石油醚/乙酸乙酯)对粗品进行纯化,得到产物苯甲酰苯乙酮。相应的产率结果见表1。
[0100]
表1不同催化剂催化合成取代酮类化合物的结果
[0101]
催化剂产率(%)催化剂a0催化剂b13triapdcl@go催化剂(实施例1)91
[0102]
虽然本发明已以较佳实施例公开如上,但其并非用以限定本发明,任何熟悉此技术的人,在不脱离本发明的精神和范围内,都可做各种的改动与修饰,因此本发明的保护范围应该以权利要求书所界定的为准。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1