一种偶氮苯二甲酰胺基桥联β-环糊精手性固定相的制备方法及其用途
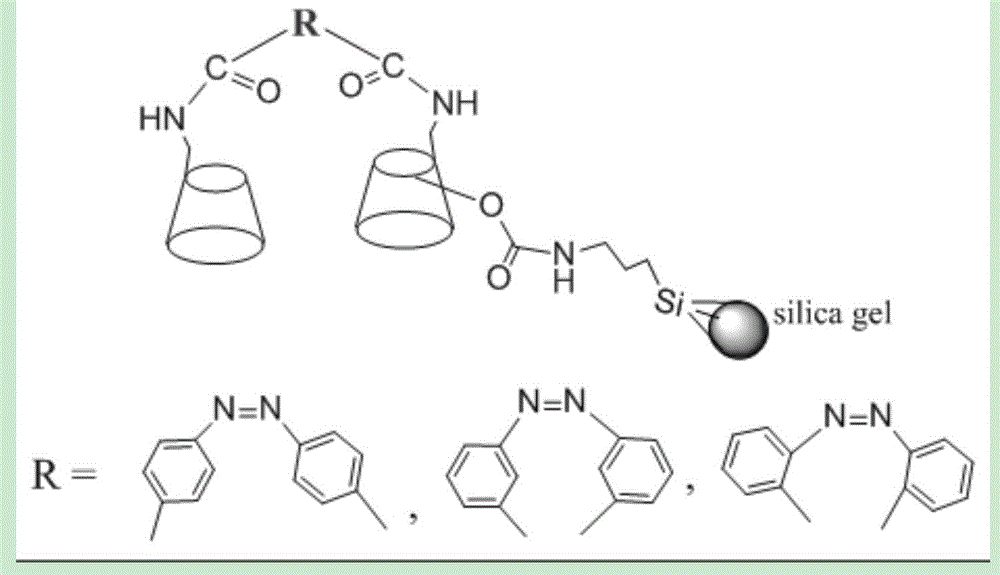
本发明涉及手性固定相技术领域,具体涉及一种偶氮苯二甲酰胺基桥联β-环糊精手性固定相的制备方法及其用途。
背景技术:
手性是自然界中常见的现象,手性化合物在化学、医学、食品、农药等方面应用广泛。基于权威机构研究表明在1800种农药中,57%的药物活性成分是手性的。由对映体引起的手性问题在20世纪初就引起了国际社会的广泛关注,“沙利度胺事件”血的教训使人们开始警醒,逐渐认识到手性物质虽然对映体的物理和化学性质几乎相同,但它们与自然界生命物质作用时可能表现出完全不同的生物活性、毒性、毒理学、代谢途径和药用价值。1992年美国fda开始要求手性药物以单一对映体的形式上市,这样不仅药品产量增长一倍、临床用量少,而且疗效更确切,副作用小。但是由于手性物质的结构非常相似,手性拆分常常遇到困难。手性拆分的关键在于手性固定相的材料,多模式的手性固定相的制备与应用一直都是分离分析中的热点研究领域。因此,建立有效实用的手性固定相和拆分方法对食品安全、药品安全、环境安全都具有重要意义。
目前拆分对映体最有效和方便的的方法是膜分离技术和色谱技术等,其中高效液相色谱法(hplc)是最有效的手性分离方法之一,hplc的分离条件温和,不会导致样品对映异构体的转变,对药品的兼容性好,得到飞速发展。高效液相色谱的分离原理主要是基于手性物质与手性固定相和流动相的作用差异性来分离的,手性物质的是否能分开,以及其分离度与色谱柱的关系最为紧密。常用的手性分离材料主要包括:刷型类、蛋白质类、大环抗生素类、纤维素类、环糊精类和金属有机框架固定相等。环糊精类固定相由于成本较低,无毒性,又适用于多种色谱分离模式,在手性药物、农药分析等众多领域得到了广泛应用,在手性分离中部分被商品化。
环糊精是由多个d-葡萄糖组成的低聚物,常见的有α-,β-,γ-环糊精(n=6,7,8),它们有类似水桶的形状,中间是可包合的中空结构,碳氧骨架,多个极性的羟基规整地排列在两个端口,形成了内疏水的空腔,外端口亲水的结构,分子内包含多个手性位点,具有较强的空间识别本领,这为手性分离创造了良好的条件,作为超分子客体已在人工仿酶、分子识别、药物载体和色谱分离等方面得到了广泛的应用。前人通过对环糊精外围端口进行了一系列的部分衍生化和全衍生化,如:甲基化、羟丙基化、苯甲酰化、苯或萘氨基甲酸酯化等,将氢键作用、静电作用,偶极-偶极作用、π-π作用等多重作用引入环糊精端口,有助于包结作用的发生,从而大大增强了手性分离能力。然而端口的衍生化,一定程度上使端口基团变得拥挤,不便于充分发挥空腔的包结作用。为此,本专利方法设计合成的桥联环糊精,在一定程度上能够弥补上述不足,通过桥基将两个或以上环糊精空腔构成一个整体,具有协同包结作用,协同包结的“假空腔”还可识别体积大的手性物质,加之桥基的协同作用,使手性识别能力进一步增强,充分发挥天然环糊精的优势。
桥联环糊精是由两个及以上的单环糊精通过间隔臂连接起来,是新一代超分子化合物。两个相邻的环糊精空腔,有可能协同参与对形状和尺寸适合的客体分子的包结配位作用,从而扩展分子的键合能力,给出最高的超分子配合物结合常数(>1011l/mol),有利于增大左旋体和右旋体与桥联环糊精结合常数的差异,从而显示出更高的手性选择性,同时克服了单一环糊精空腔较小(~0.65nm)的不足,可分离分子体积更大的手性化合物。
桥基的选择也很重要,偶氮苯二甲酰基桥基含有大π-共轭体系、氢键提供和接受体,特别是偶氮基顺反转换能垒较低,这种柔性的桥基可更好地配合两个相邻环糊精空腔协同包结作用。本专利方法设计以单-6-氨基-β-环糊精为中间体、偶氮苯-4,4'-二甲酰基基为桥联基团,合成了偶氮苯二甲酰胺基桥联β-环糊精手性配体,然后以3-异氰酸丙基三乙氧基硅烷为偶联剂,以有序介孔sba-15硅胶为基质,制备得到新型的偶氮苯二甲酰胺基桥联β-环糊精手性固定相。柔性的偶氮苯-4,4'-二甲酰基作为桥基与两个环糊精分子构成钳式结构,有利于协同包结并识别手性化合物。另外,有序介孔sba-15硅胶作基质,固定相孔道规整,涡流扩散小,传质速度快,有利于快速分析。
本发明方法合成的偶氮苯二甲酰胺基桥联β-环糊精键合sba-15手性固定相在手性物质分离中具有明显的优势,对于传统的β-环糊精固定相很难拆分的手性化合物:3'-羟基黄烷酮(a)、丹磺酰亮氨酸(b)、卡维地洛(c)、烯效唑(d),可在偶氮苯二甲酰胺基桥联β-环糊精手性固定相上短时间内完全被拆分。
技术实现要素:
本发明目的是提供一种偶氮苯二甲酰胺基桥联β-环糊精键合sba-15手性固定相的制备方法及其在分离手性化合物方面的用途。
为实现上述目的,本发明提供一种偶氮苯二甲酰胺基桥联β-环糊精键合手性固定相,所手性固定相制备方法包括以下步骤:
(1)将偶氮苯二甲酸,单-6-氨基-β-环糊精,n,n’-二环己基碳二亚胺,1-羟基苯并三唑和n,n-二甲基甲酰胺混合,常温下磁力搅拌反应48h,得到偶氮苯二甲酰胺基桥联β-环糊精的溶液,将丙酮加入上述反应液后析出沉淀,过滤,固体用水溶解,通过羧甲基葡聚糖凝胶c-25柱分离纯化,洗脱液加丙酮析出产物,干燥,得到偶氮苯二甲酰胺基桥联β-环糊精手性配体;
(2)在氮气保护下,将偶氮苯二甲酰胺基桥联β-环糊精溶于无水n,n-二甲基甲酰胺,缓慢加入0.2-0.5ml的γ-异氰酸基丙基三乙氧基硅烷,80℃磁力搅拌反应2h,得到含偶氮苯二甲酰胺基桥联β-环糊精的三乙氧基硅烷反应液;
(3)在氮气保护下,加入偶氮苯二甲酰胺基桥联β-环糊精:介孔分子筛sba-15比例为1:3.5的干燥的sba-15到步骤(2)的反应液中,然后升温至105~125℃反应12h,过滤,固体用n,n-二甲基甲酰胺、甲醇和水反复洗涤,真空干燥,得到偶氮苯二甲酰胺基桥联β-环糊精键合sba-15手性固定相粗产品;
(4)以偶氮苯二甲酰胺基桥联β-环糊精键合sba-15的手性固定相粗产品:丙酮为1:100的比例,加入丙酮提取,索氏提取12h以除去粗产品腔体和孔隙中未反应试剂,然后在50℃真空干燥8h,得到偶氮苯二甲酰胺基桥联β-环糊精键合sba-15手性固定相产品。
优选的,所述步骤(1)中,偶氮苯二甲酸:单-6-氨基-β-环糊精:n,n’-二环己基碳二亚胺:1-羟基苯并三唑:n,n-二甲基甲酰胺的比例为1:1.5~3.0:1.5~3.0:1.5~3.0:15~30。
优选的,所述比例以偶氮苯二甲酸:单-6-氨基-β-环糊精:n,n’-二环己基碳二亚胺:1-羟基苯并三唑:n,n-二甲基甲酰胺为1.0:2.0:2.0:2.0:20为最优。
优选的,所述步骤(1)中偶氮苯二甲酸为偶氮苯-4,4'-二甲酸、偶氮苯-3,3'-二甲酸或偶氮苯-2,2'-二甲酸中的一种。
优选的,所述步骤(2)中γ-异氰酸基丙基三乙氧基硅烷用量以0.4ml为最优。
优选的,所述步骤(3)中反应温度以115℃为最优。
优选的,所述偶氮苯二甲酰胺基桥联β-环糊精手性固定相用于3'-羟基黄烷酮、丹磺酰亮氨酸、卡维地洛、烯效唑手性化合物对映体拆分。
本发明的有益效果是:
(1)本发明以3'-羟基黄烷酮、丹磺酰亮氨酸、卡维地洛、烯效唑为手性溶质探针,在多种模式下评价该固定相的手性色谱性能,得到结果:未衍生化的偶氮苯二甲酰胺基桥联β-环糊精手性固定相对对映体在短时间内能达到较好的分离度,桥联双环糊精具有协同识别能力。
(2)新固定相环糊精端口保留了大量的羟基,具有较强的氢键作用,苯环提供π-π作用、酰胺既是氢键给体又是氢键受体、偶氮是柔性结构,分子整体呈现钳式结构,在手性物质拆分过程中能协同包结被分析物,增大了对映体与环糊精形成包合物稳定性的差异,为手性物质的拆分提供更多可能性。
(3)偶氮苯二甲酰胺基桥联β-环糊精键合sba-15手性固定相制备方法具有流程简单、原材料成本低廉、溶质选择性好、适用于多种色谱模式,色谱性能稳定、重现性好等优点。
附图说明
图1为偶氮苯二甲酰胺基桥联β-环糊精手性固定相的结构示意图;
图2为所拆分的手性化合物的化学结构;
图3为3'-羟基黄烷酮(a)、丹磺酰亮氨酸(b)、卡维地洛(c)、烯效唑(d)的手性物质分离色谱图。
具体实施方式
下面结合实例对本发明进行进一步的说明。
实施例1
(1)将偶氮苯-4,4’-二甲酸(mmol):单-6-氨基-β-环糊精(mmol):n,n’-二环己基碳二亚胺(dcc,mmol):1-羟基苯并三唑(hobt,mmol):n,n-二甲基甲酰胺(dmf,ml)按1.0:1.5:1.5:1.5:15的比例混合,常温下反应48h,得到偶氮苯二甲酰胺基桥联β-环糊精的溶液,将丙酮加入上述反应液后析出沉淀,过滤,固体用水溶解,通过羧甲基葡聚糖凝胶(c-25)柱分离纯化,洗脱液加丙酮析出产物,干燥,得到偶氮苯-4,4’-二甲酰胺基桥联β-环糊精手性配体;
(2)在氮气保护下,将偶氮苯二甲酰胺基桥联β-环糊精溶于无水dmf,然后将0.4ml的γ-异氰酸基丙基三乙氧基硅烷缓慢加入上述溶液,于80℃反应2h,得到含偶氮苯-4,4’-二甲酰胺基桥联β-环糊精三乙氧基硅烷的反应液;
(3)在氮气保护下,按(2)中的偶氮苯二甲酰胺基桥联β-环糊精(mmol):sba-15(g)为1:3.5的比例,将干燥的sba-15直接加入(2)步的反应液中,然后升温至115℃反应12h,过滤,固定用dmf、甲醇和水反复洗涤,干燥,得到偶氮苯-4,4’-二甲酰胺基桥联β-环糊精键合sba-15的手性固定相粗产品;
(4)按(3)合成的偶氮苯-4,4’-二甲酰胺基桥联β-环糊精键合相(g):丙酮(ml)为1:100的比例,以丙酮为提取剂,索氏提取12h以除去粗产品腔体和孔隙中未反应的试剂,然后在50℃真空干燥8h,得到偶氮苯二甲酰胺基桥联β-环糊精键合sba-15的手性固定相产品。
通过元素分析结果中的碳含量计算所制得的偶氮苯-4,4’-二甲酰胺基桥联β-环糊精键合sba-15手性固定相的键合量,实测数据如下表:
实施例2
(1)将偶氮苯-4,4’-二甲酸(mmol):单-6-氨基-β-环糊精(mmol):dcc(mmol):hobt(mmol):dmf(ml)按1.0:2.0:2.0:2.0:20的比例混合,常温下反应48h,得到偶氮苯二甲酰胺基桥联β-环糊精的溶液,将丙酮加入上述反应液后析出沉淀,过滤,固体用水溶解,通过羧甲基葡聚糖凝胶(c-25)柱分离纯化,洗脱液加丙酮析出产物,干燥,得到偶氮苯-4,4’-二甲酰胺基桥联β-环糊精手性配体;
(2)在氮气保护下,将偶氮苯二甲酰胺基桥联β-环糊精溶于无水dmf,然后将0.4ml的γ-异氰酸基丙基三乙氧基硅烷缓慢加入上述溶液,于80℃反应2h,得到含偶氮苯-4,4’-二甲酰胺基桥联β-环糊精三乙氧基硅烷的反应液;
(3)在氮气保护下,按(2)中的偶氮苯二甲酰胺基桥联β-环糊精(mmol):sba-15(g)为1:3.5的比例,将干燥的sba-15直接加入(2)步的反应液中,然后升温至115℃反应12h,过滤,固定用dmf、甲醇和水反复洗涤,干燥,得到偶氮苯-4,4’-二甲酰胺基桥联β-环糊精键合sba-15的手性固定相粗产品;
(4)按(3)合成的偶氮苯-4,4’-二甲酰胺基桥联β-环糊精键合相(g):丙酮(ml)为1:100的比例,以丙酮为提取剂,索氏提取12h以除去粗产品腔体和孔隙中未反应的试剂,然后在50℃真空干燥8h,得到偶氮苯二甲酰胺基桥联β-环糊精键合sba-15的手性固定相产品。
通过元素分析结果中的碳含量计算所制得的偶氮苯-4,4’-二甲酰胺基桥联β-环糊精键合sba-15手性固定相的键合量,实测数据如下表:
实施例3
(1)将偶氮苯-4,4’-二甲酸(mmol):单-6-氨基-β-环糊精(mmol):dcc(mmol):hobt(mmol):dmf(ml)按1.0:2.5:2.5:2.5:25的比例混合,常温下反应48h,得到偶氮苯二甲酰胺基桥联β-环糊精的溶液,将丙酮加入上述反应液后析出沉淀,过滤,固体用水溶解,通过羧甲基葡聚糖凝胶(c-25)柱分离纯化,洗脱液加丙酮析出产物,干燥,得到偶氮苯-4,4’-二甲酰胺基桥联β-环糊精手性配体;
(2)在氮气保护下,将偶氮苯二甲酰胺基桥联β-环糊精溶于无水dmf,然后将0.4ml的γ-异氰酸基丙基三乙氧基硅烷缓慢加入上述溶液,于80℃反应2h,得到含偶氮苯-4,4’-二甲酰胺基桥联β-环糊精三乙氧基硅烷的反应液;
(3)在氮气保护下,按(2)中的偶氮苯二甲酰胺基桥联β-环糊精(mmol):sba-15(g)为1:3.5的比例,将干燥的sba-15直接加入(2)步的反应液中,然后升温至115℃反应12h,过滤,固定用dmf、甲醇和水反复洗涤,干燥,得到偶氮苯-4,4’-二甲酰胺基桥联β-环糊精键合sba-15的手性固定相粗产品;
(4)按(3)合成的偶氮苯-4,4’-二甲酰胺基桥联β-环糊精键合相(g):丙酮(ml)为1:100的比例,以丙酮为提取剂,索氏提取12h以除去粗产品腔体和孔隙中未反应的试剂,然后在50℃真空干燥8h,得到偶氮苯二甲酰胺基桥联β-环糊精键合sba-15的手性固定相产品。
本专利通过元素分析结果中的碳含量计算所制得的偶氮苯-4,4’-二甲酰胺基桥联β-环糊精键合sba-15手性固定相的键合量,实测数据如下表:
实施例4
(1)将偶氮苯-4,4’-二甲酸(mmol):单-6-氨基-β-环糊精(mmol):dcc(mmol):hobt(mmol):dmf(ml)按1.0:3.0:3.0:3.0:30的比例混合,常温下反应48h,得到偶氮苯二甲酰胺基桥联β-环糊精的溶液,将丙酮加入上述反应液后析出沉淀,过滤,固体用水溶解,通过羧甲基葡聚糖凝胶(c-25)柱分离纯化,洗脱液加丙酮析出产物,干燥,得到偶氮苯-4,4’-二甲酰胺基桥联β-环糊精手性配体;
(2)在氮气保护下,将偶氮苯二甲酰胺基桥联β-环糊精溶于无水dmf,然后将0.4ml的γ-异氰酸基丙基三乙氧基硅烷缓慢加入上述溶液,于80℃反应2h,得到含偶氮苯-4,4’-二甲酰胺基桥联β-环糊精三乙氧基硅烷的反应液;
(3)在氮气保护下,按(2)中的偶氮苯二甲酰胺基桥联β-环糊精(mmol):sba-15(g)为1:3.5的比例,将干燥的sba-15直接加入(2)步的反应液中,然后升温至115℃反应12h,过滤,固定用dmf、甲醇和水反复洗涤,干燥,得到偶氮苯-4,4’-二甲酰胺基桥联β-环糊精键合sba-15的手性固定相粗产品;
(4)按(3)合成的偶氮苯-4,4’-二甲酰胺基桥联β-环糊精键合相(g):丙酮(ml)为1:100的比例,以丙酮为提取剂,索氏提取12h以除去粗产品腔体和孔隙中未反应的试剂,然后在50℃真空干燥8h,得到偶氮苯二甲酰胺基桥联β-环糊精键合sba-15的手性固定相产品。
本专利通过元素分析结果中的碳含量计算所制得的偶氮苯-4,4’-二甲酰胺基桥联β-环糊精键合sba-15手性固定相的键合量,实测数据如下表:
实施例5
(1)将偶氮苯-3,3’-二甲酸(mmol):单-6-氨基-β-环糊精(mmol):dcc(mmol):hobt(mmol):dmf(ml)按1.0:2.0:2.0:2.0:20的比例混合,常温下反应48h,得到偶氮苯二甲酰胺基桥联β-环糊精的溶液,将丙酮加入上述反应液后析出沉淀,过滤,固体用水溶解,通过羧甲基葡聚糖凝胶(c-25)柱分离纯化,洗脱液加丙酮析出产物,干燥,得到偶氮苯-3,3’-二甲酰胺基桥联β-环糊精手性配体;
(2)在氮气保护下,将偶氮苯二甲酰胺基桥联β-环糊精溶于无水dmf,然后将0.4ml的γ-异氰酸基丙基三乙氧基硅烷缓慢加入上述溶液,于80℃反应2h,得到含偶氮苯-3,3’-二甲酰胺基桥联β-环糊精三乙氧基硅烷的反应液;
(3)在氮气保护下,按(2)中的偶氮苯二甲酰胺基桥联β-环糊精(mmol):sba-15(g)为1:3.5的比例,将干燥的sba-15直接加入(2)步的反应液中,然后升温至115℃反应12h,过滤,固定用dmf、甲醇和水反复洗涤,干燥,得到偶氮苯-3,3’-二甲酰胺基桥联β-环糊精键合sba-15的手性固定相粗产品;
(4)按(3)合成的偶氮苯-3,3’-二甲酰胺基桥联β-环糊精键合相(g):丙酮(ml)为1:100的比例,以丙酮为提取剂,索氏提取12h以除去粗产品腔体和孔隙中未反应的试剂,然后在50℃真空干燥8h,得到偶氮苯二甲酰胺基桥联β-环糊精键合sba-15的手性固定相产品。
通过元素分析结果中的碳含量计算所制得的偶氮苯-3,3’-二甲酰胺基桥联β-环糊精键合sba-15手性固定相的键合量,实测数据如下表:
实施例6
(1)将偶氮苯-2,2’-二甲酸(mmol):单-6-氨基-β-环糊精(mmol):dcc(mmol):hobt(mmol):dmf(ml)按1.0:2.0:2.0:2.0:20的比例混合,常温下反应48h,得到偶氮苯二甲酰胺基桥联β-环糊精的溶液,将丙酮加入上述反应液后析出沉淀,过滤,固体用水溶解,通过羧甲基葡聚糖凝胶(c-25)柱分离纯化,洗脱液加丙酮析出产物,干燥,得到偶氮苯-2,2’-二甲酰胺基桥联β-环糊精手性配体;
(2)在氮气保护下,将偶氮苯二甲酰胺基桥联β-环糊精溶于无水dmf,然后将0.4ml的γ-异氰酸基丙基三乙氧基硅烷缓慢加入上述溶液,于80℃反应2h,得到含偶氮苯-2,2’-二甲酰胺基桥联β-环糊精三乙氧基硅烷的反应液;
(3)在氮气保护下,按(2)中的偶氮苯二甲酰胺基桥联β-环糊精(mmol):sba-15(g)为1:3.5的比例,将干燥的sba-15直接加入(2)步的反应液中,然后升温至115℃反应12h,过滤,固定用dmf、甲醇和水反复洗涤,干燥,得到偶氮苯-2,2’-二甲酰胺基桥联β-环糊精键合sba-15的手性固定相粗产品;
(4)按(3)合成的偶氮苯-2,2’-二甲酰胺基桥联β-环糊精键合相(g):丙酮(ml)为1:100的比例,以丙酮为提取剂,索氏提取12h以除去粗产品腔体和孔隙中未反应的试剂,然后在50℃真空干燥8h,得到偶氮苯二甲酰胺基桥联β-环糊精键合sba-15的手性固定相产品。
本专利通过元素分析结果中的碳含量计算所制得的偶氮苯-2,2’-二甲酰胺基桥联β-环糊精键合sba-15手性固定相的键合量,实测数据如下表:
实施例7
(1)将偶氮苯-4,4’-二甲酸(mmol):单-6-氨基-β-环糊精(mmol):dcc(mmol):hobt(mmol):dmf(ml)按1.0:2.0:2.0:2.0:20的比例混合,常温下反应48h,得到偶氮苯二甲酰胺基桥联β-环糊精的溶液,将丙酮加入上述反应液后析出沉淀,过滤,固体用水溶解,通过羧甲基葡聚糖凝胶(c-25)柱分离纯化,洗脱液加丙酮析出产物,干燥,得到偶氮苯-4,4’-二甲酰胺基桥联β-环糊精手性配体;
(2)在氮气保护下,将偶氮苯二甲酰胺基桥联β-环糊精溶于无水dmf,然后将0.4ml的γ-异氰酸基丙基三乙氧基硅烷缓慢加入上述溶液,于80℃反应2h,得到含偶氮苯-4,4’-二甲酰胺基桥联β-环糊精三乙氧基硅烷的反应液;
(3)在氮气保护下,按(2)中的偶氮苯二甲酰胺基桥联β-环糊精(mmol):sba-15(g)为1:3.5的比例,将干燥的sba-15直接加入(2)步的反应液中,然后升温至105℃反应12h,过滤,固定用dmf、甲醇和水反复洗涤,干燥,得到偶氮苯-4,4’-二甲酰胺基桥联β-环糊精键合sba-15的手性固定相粗产品;
(4)按(3)合成的偶氮苯-4,4’-二甲酰胺基桥联β-环糊精键合相(g):丙酮(ml)为1:100的比例,以丙酮为提取剂,索氏提取12h以除去粗产品腔体和孔隙中未反应的试剂,然后在50℃真空干燥8h,得到偶氮苯二甲酰胺基桥联β-环糊精键合sba-15的手性固定相产品。
通过元素分析结果中的碳含量计算所制得的偶氮苯-4,4’-二甲酰胺基桥联β-环糊精键合sba-15手性固定相的键合量,实测数据如下表:
实施例8
(1)将偶氮苯-4,4’-二甲酸(mmol):单-6-氨基-β-环糊精(mmol):dcc(mmol):hobt(mmol):dmf(ml)按1.0:2.0:2.0:2.0:20的比例混合,常温下反应48h,得到偶氮苯二甲酰胺基桥联β-环糊精的溶液,将丙酮加入上述反应液后析出沉淀,过滤,固体用水溶解,通过羧甲基葡聚糖凝胶(c-25)柱分离纯化,洗脱液加丙酮析出产物,干燥,得到偶氮苯-4,4’-二甲酰胺基桥联β-环糊精手性配体;
(2)在氮气保护下,将偶氮苯二甲酰胺基桥联β-环糊精溶于无水dmf,然后将0.4ml的γ-异氰酸基丙基三乙氧基硅烷缓慢加入上述溶液,于80℃反应2h,得到含偶氮苯-4,4’-二甲酰胺基桥联β-环糊精三乙氧基硅烷的反应液;
(3)在氮气保护下,按(2)中的偶氮苯二甲酰胺基桥联β-环糊精(mmol):sba-15(g)为1:3.5的比例,将干燥的sba-15直接加入(2)步的反应液中,然后升温至125℃反应12h,过滤,固定用dmf、甲醇和水反复洗涤,干燥,得到偶氮苯-4,4’-二甲酰胺基桥联β-环糊精键合sba-15的手性固定相粗产品;
(4)按(3)合成的偶氮苯-4,4’-二甲酰胺基桥联β-环糊精键合相(g):丙酮(ml)为1:100的比例,以丙酮为提取剂,索氏提取12h以除去粗产品腔体和孔隙中未反应的试剂,然后在50℃真空干燥8h,得到偶氮苯二甲酰胺基桥联β-环糊精键合sba-15的手性固定相产品。
本专利通过元素分析结果中的碳含量计算所制得的偶氮苯-4,4’-二甲酰胺基桥联β-环糊精键合sba-15手性固定相的键合量,实测数据如下表:
实施例9
(1)将偶氮苯-4,4’-二甲酸(mmol):单-6-氨基-β-环糊精(mmol):dcc(mmol):hobt(mmol):dmf(ml)按1.0:2.0:2.0:2.0:20的比例混合,常温下反应48h,得到偶氮苯二甲酰胺基桥联β-环糊精的溶液,将丙酮加入上述反应液后析出沉淀,过滤,固体用水溶解,通过羧甲基葡聚糖凝胶(c-25)柱分离纯化,洗脱液加丙酮析出产物,干燥,得到偶氮苯-4,4’-二甲酰胺基桥联β-环糊精手性配体;
(2)在氮气保护下,将偶氮苯二甲酰胺基桥联β-环糊精溶于无水dmf,然后将0.2ml的γ-异氰酸基丙基三乙氧基硅烷缓慢加入上述溶液,于80℃反应2h,得到含偶氮苯-4,4’-二甲酰胺基桥联β-环糊精三乙氧基硅烷的反应液;
(3)在氮气保护下,按(2)中的偶氮苯二甲酰胺基桥联β-环糊精(mmol):sba-15(g)为1:3.5的比例,将干燥的sba-15直接加入(2)步的反应液中,然后升温至115℃反应12h,过滤,固定用dmf、甲醇和水反复洗涤,干燥,得到偶氮苯-4,4’-二甲酰胺基桥联β-环糊精键合sba-15的手性固定相粗产品;
(4)按(3)合成的偶氮苯-4,4’-二甲酰胺基桥联β-环糊精键合相(g):丙酮(ml)为1:100的比例,以丙酮为提取剂,索氏提取12h以除去粗产品腔体和孔隙中未反应的试剂,然后在50℃真空干燥8h,得到偶氮苯二甲酰胺基桥联β-环糊精键合sba-15的手性固定相产品。
本专利通过元素分析结果中的碳含量计算所制得的偶氮苯-4,4’-二甲酰胺基桥联β-环糊精键合sba-15手性固定相的键合量,实测数据如下表:
实施例10
(1)将偶氮苯-4,4’-二甲酸(mmol):单-6-氨基-β-环糊精(mmol):dcc(mmol):hobt(mmol):dmf(ml)按1.0:2.0:2.0:2.0:20的比例混合,常温下反应48h,得到偶氮苯二甲酰胺基桥联β-环糊精的溶液,将丙酮加入上述反应液后析出沉淀,过滤,固体用水溶解,通过羧甲基葡聚糖凝胶(c-25)柱分离纯化,洗脱液加丙酮析出产物,干燥,得到偶氮苯-4,4’-二甲酰胺基桥联β-环糊精手性配体;
(2)在氮气保护下,将偶氮苯二甲酰胺基桥联β-环糊精溶于无水dmf,然后将0.3ml的γ-异氰酸基丙基三乙氧基硅烷缓慢加入上述溶液,于80℃反应2h,得到含偶氮苯-4,4’-二甲酰胺基桥联β-环糊精三乙氧基硅烷的反应液;
(3)在氮气保护下,按(2)中的偶氮苯二甲酰胺基桥联β-环糊精(mmol):sba-15(g)为1:3.5的比例,将干燥的sba-15直接加入(2)步的反应液中,然后升温至115℃反应12h,过滤,固定用dmf、甲醇和水反复洗涤,干燥,得到偶氮苯-4,4’-二甲酰胺基桥联β-环糊精键合sba-15的手性固定相粗产品;
(4)按(3)合成的偶氮苯-4,4’-二甲酰胺基桥联β-环糊精键合相(g):丙酮(ml)为1:100的比例,以丙酮为提取剂,索氏提取12h以除去粗产品腔体和孔隙中未反应的试剂,然后在50℃真空干燥8h,得到偶氮苯二甲酰胺基桥联β-环糊精键合sba-15的手性固定相产品。
通过元素分析结果中的碳含量计算所制得的偶氮苯-4,4’-二甲酰胺基桥联β-环糊精键合sba-15手性固定相的键合量,实测数据如下表:
实施例11
(1)将偶氮苯-4,4’-二甲酸(mmol):单-6-氨基-β-环糊精(mmol):dcc(mmol):hobt(mmol):dmf(ml)按1.0:2.0:2.0:2.0:20的比例混合,常温下反应48h,得到偶氮苯二甲酰胺基桥联β-环糊精的溶液,将丙酮加入上述反应液后析出沉淀,过滤,固体用水溶解,通过羧甲基葡聚糖凝胶(c-25)柱分离纯化,洗脱液加丙酮析出产物,干燥,得到偶氮苯-4,4’-二甲酰胺基桥联β-环糊精手性配体;
(2)在氮气保护下,将偶氮苯二甲酰胺基桥联β-环糊精溶于无水dmf,然后将0.5ml的γ-异氰酸基丙基三乙氧基硅烷缓慢加入上述溶液,于80℃反应2h,得到含偶氮苯-4,4’-二甲酰胺基桥联β-环糊精三乙氧基硅烷的反应液;
(3)在氮气保护下,按(2)中的偶氮苯二甲酰胺基桥联β-环糊精(mmol):sba-15(g)为1:3.5的比例,将干燥的sba-15直接加入(2)步的反应液中,然后升温至115℃反应12h,过滤,固定用dmf、甲醇和水反复洗涤,干燥,得到偶氮苯-4,4’-二甲酰胺基桥联β-环糊精键合sba-15的手性固定相粗产品;
(4)按(3)合成的偶氮苯-4,4’-二甲酰胺基桥联β-环糊精键合相(g):丙酮(ml)为1:100的比例,以丙酮为提取剂,索氏提取12h以除去粗产品腔体和孔隙中未反应的试剂,然后在50℃真空干燥8h,得到偶氮苯二甲酰胺基桥联β-环糊精键合sba-15的手性固定相产品。
本专利通过元素分析结果中的碳含量计算所制得的偶氮苯-4,4’-二甲酰胺基桥联β-环糊精键合sba-15手性固定相的键合量,实测数据如下表:
实施例12
高效液相色谱手性分离实验
分别将实施例2、实施例5、实施例6所制备的偶氮苯-4,4’-二甲酰胺基桥联β-环糊精手性固定相、偶氮苯-3,3’-二甲酰胺基桥联β-环糊精手性固定相、偶氮苯-2,2'-二甲酰胺基桥联β-环糊精手性固定相作为高效液相色谱柱填料,对一些手性化合物进行分离。
拆分的手性物质:3’-羟基黄烷酮、丹磺酰亮氨酸、卡维地洛、烯效唑。
具体步骤:采用匀浆法进行色谱柱的装填。其中,匀浆剂为丙酮;顶替剂为甲醇。将匀浆液填装进不锈钢色谱柱(4.6mm×250mm),维持35.0mpa的压力30分钟,然后逐渐卸压以完成装柱,100%乙腈、100%甲醇、不同体积比的水和甲醇作为流动相在低流速下各冲洗半小时,直至基线平稳后开始进样,所有标准品用甲醇溶解,配制成100~200μg/ml的储备液,于4℃避光存放。进样前用0.22μm滤膜过滤并超声脱气。反相色谱模式以甲醇/水或乙腈/水为流动相,流速为0.8ml/min;柱温为20℃;进样量为5μl;检测波长范围200~380nm。极性有机模式以适当比的甲醇/乙腈/三乙胺/乙酸为流动相。其余色谱参数与反相色谱模式相同。
实验结果及其相应优化的色谱条件见图,可以看出,上述溶质均被完全分离。显然,本发明偶氮苯二甲酰胺基桥联β-环糊精手性固定相能被用于高效液相色谱法拆分部分手性药物如黄烷酮类、氨基酸类、β-受体阻滞剂等药物和三唑类手性农药的对映体。
- 还没有人留言评论。精彩留言会获得点赞!