产品中5种松香酸的高效液相色谱串联质谱的分析方法与流程
本发明属于分析检测技术领域,具体涉及一种产品中5种松香酸的高效液相色谱串联质谱的分析方法。
背景技术:
松香具有粘合、防腐、绝缘等优良的特性,被广泛地应用在胶粘剂、塑料、涂料、油墨、造纸和医药等领域。在日常的消费品中,松香可能存在肥皂、纸张、涂料以及使用胶黏剂和塑料的各种鞋、玩具等产品。松香被大量使用的同时,其危害性也逐渐被人们发现和关注。松香浓蒸汽可引起头疼、昏眩、咳嗽、气喘等急性中毒症状。曾有报导指出,树脂酸对鱼的毒性为0.44μg/mL。毒理学研究表明松香酸可导致人体肺泡上皮细胞溶解,脱氢松香酸对人体红细胞、多核白细胞有毒性作用,Asako Ozaki等发现脱氢松香酸和松香酸具有损伤DNA的活性,Yasushi Kamaya等研究了松香酸、脱氢松香酸对大型水蚤生长的影响,发现二者对水蚤的生长有一定的限制作用。研究表明,松香是全球十大皮肤过敏源物质,长时间暴露在焊接释放出的松香烟雾可引起职业性哮喘,根据欧盟的分类法规,松香大于1%的产品将标识为R43(即能引起皮肤过敏)。
松香是一种无定形透明玻璃状固体树脂,主要成分是树脂酸,占总质量85%~90%,另外还有少量的脂肪酸和中性物质。目前国内外研究报道的对人体过敏的松香主要为枞酸(abietic acid)、新枞酸(Neoabietic acid)、左旋海松酸、海松酸(Pimaric acid)和脱氢枞酸(Dehydroabietic acid)5种。5种松香酸的CAS号、结构式如下所示:
有关松香酸含量的检测技术研究包括松香产品质量分析以及某些产品中松香酸的含量分析。对于松香品质的鉴定,已有国家标准,但该标准不涉及松香中各树脂酸的测定。魏福祥等采用红外光谱法定量测定了弹性体以及松香、液体石蜡混合物中松香酸的含量,但该方法检出限低,受基体干扰大,不能满足各类消费品中松香酸含量测定的要求。由于松香酸的毒性作用,某些产品中掺入或残留的松香酸含量的检测方法研究也引起了人们关注。对于松香酸中各种树脂酸含量的测定,通常可采用薄层色谱法、气相色谱法、气质联用法、高效液相色谱法、液质联用法等方法。这些方法存在两个方面的不足:一、检测方法只涉及松香酸中的枞酸和脱氢松香酸两种物质,而松香酸包含多种物质,检测覆盖面不足;二,松香酸广泛存在于各种日常消费品中,如鞋材中的胶黏剂、纸张、松香皂、油墨等。而报道的文献中主要集中在沉香、松香及纸制品中。为了保护消费者的身体健康和生命安全,应对国外贸易技术壁垒,提前做好技术储备工作,保证产品正常出口,维护社会稳定促进经济社会建设具有积极作用,有必要制定产品中松香酸的检测方法。
CN201310144917公开一种畜禽表皮中松香酸含量的分析方法,涉及畜禽表皮中松香酸的分析方法,属于分析测试领域。在屠宰加工后的生畜禽的表皮样品中加入稀碱溶液,经提取、固相萃取净化后,取得滤过液,再将滤过液采用高效液相色谱-紫外法(HPLC-UV)进行分析。该方法无法检测分离5种松香酸,具有一定的局限性。
技术实现要素:
针对现有技术的不足,本发明提供一种产品中5种松香酸的高效液相色谱串联质谱的分析方法,该方法采用高效液相色谱串联质谱的方法测定4类产品中5种松香酸的含量;该方法优化了液相色谱的流动相,检测步骤少,测试结果准确,能快速的检测出松香酸的含量,抗干扰性强,能有效检测5种松香酸。
为解决上述技术问题,本发明提供如下技术方案:
产品中5种松香酸的高效液相色谱串联质谱的分析方法,其特征在于,具体检测步骤为:i)样品的处理:称取待测样品,置于管状硬质玻璃提取器中,加入第一提取溶剂,第一次超声提取,过滤;样品中再加入第二提取溶剂,第二次超声提取,过滤,将两次过滤的滤液合并后进行浓缩,加入定容溶剂溶解后在容量瓶中定容,取样品溶液经有机滤膜过滤后,提取液待用;
ii)将步骤i)所得提取液用HPLC-MS进行测定,选择两级质谱的特定离子对,比较试样和标样色谱峰的保留时间进行定性,以峰面积外标法定量。
进一步地,所述高效液相色谱检测的液相色谱条件为:
液相色谱柱:Waters Atlantis T3,100×2.1mm,3μm;
流动相:乙腈和0.3%乙酸水溶液的混合溶液,流量为0.3mL·min-1;
进样量为5uL,波长为200nm、240nm、280nm,柱温40℃。
进一步地,所述质谱检测的质谱条件为:
a)离子源:电喷雾离子源(ESI+);
b)离子模式:正离子模式
c)检测方式:多反应离子监测(MRM);
d)定性离子对:m/z 303.2/148.9和m/z 303.2/123.0(枞酸);m/z 303.2/148.9和m/z303.2/123.0(新枞酸);m/z 303.2/148.9和m/z 303.2/123.0(左旋海松酸);m/z 303.2/148.9和m/z 303.2/123.0(海松酸);m/z 301.2/172.9和m/z 301.2/240.0(脱氢松香酸);
j)定量离子对:m/z 303.2/257.6(枞酸);m/z 303.2/257.6(新枞酸);m/z 303.2/257.6(左旋海松酸);m/z 303.2/257.6(海松酸);m/z 301.2/172.9(脱氢松香酸);
k)干燥气温度:325℃;
l)干燥气流速:10L/min;
m)雾化气压力:40Psi;
n)电喷雾电压:4000V;
o)离子源温度:100℃;
p)毛细管出口电压和碰撞电压如下表1:
进一步地,所述容量瓶为25mL,样品称取1g。
进一步地,所述第一提取溶剂为5~15mL;所述第二提取溶剂为5~15mL。
进一步地,所述第一次超声提取和第二次超声提取的时间均为10~30min。
进一步地,所述第一次超声提取和第二次超声提取的超声波输出功率均为50~80%。
进一步地,所述第一次超声提取和第二次超声提取的温度均为40~80℃。
更进一步地,所述第一次超声提取和第二次超声提取的时间均为30min。
更进一步地,所述第一次超声提取和第二次超声提取的超声波输出功率均为80%。
更进一步地,所述第一次超声提取和第二次超声提取的温度均为60℃。
进一步地,所述待测样品为经过预处理的胶黏剂、纸张、松香皂或油墨。
更进一步地,所述胶黏剂为鞋类部件的胶黏剂、热熔胶样品、液态胶黏剂。更进一步地,所述胶黏剂为309胶黏剂,热熔胶,乳胶,以及使用胶水的鞋类部件如鞋底衬中的胶合部位、鞋面的胶合部位、鞋后跟的胶合部位,鞋底的胶合部位。更进一步地,所述纸张为瓦楞原纸、白卡纸等。更进一步地,所述油墨为快干凉胶印刷油墨。
进一步地,所述纸张包括纸制品。
进一步地,所述预处理的胶粘剂的处理过程如下:对于鞋类部件的胶黏剂,其通过将使用胶水粘合的鞋类部件,用刀刮起材料中的胶黏剂;若无法刮起,或者量不足,则直接取鞋类部件检测;
对于热熔胶样品,将样品剪成约1mm×1mm的小碎片后检测;
对于液态胶黏剂,将胶黏剂倒在玻璃平板上,自然晾干,剪成约1mm×1mm的小碎片后检测。
进一步地,所述预处理纸张的处理过程为:将纸张进行剪碎成约1mm×1mm的小碎片即可用于检测。
进一步地,所述预处理松香皂的处理过程为:将松香皂进行剪碎成约1mm×1mm的小碎片即可用于检测。
进一步地,所述预处理油墨的处理过程为:油墨材料在玻璃板上自然晾干后,剪碎;印刷制品用刮刀刮下,若无法刮下,则直接连基体材料一同处理。
进一步地,所述步骤i)中第一提取溶剂为叔丁基甲醚、水、乙腈、乙酸乙酯、二氯甲烷、丙酮、甲醇、甲醇-水的混合溶剂(V/V=75/25)、乙腈-0.3%乙酸水溶液(V/V=65/35)、乙腈-0.3%乙酸水溶液(V/V=55/45)、乙腈-0.3%乙酸水溶液(V/V=75/25)。
进一步地,所述步骤i)中第二提取溶剂为叔丁基甲醚、水、乙腈、乙酸乙酯、二氯甲烷、丙酮、甲醇、甲醇-水的混合溶剂(V/V=75/25)、乙腈-0.3%乙酸水溶液(V/V=65/35)、乙腈-0.3%乙酸水溶液(V/V=55/45)、乙腈-0.3%乙酸水溶液(V/V=75/25)。
进一步地,所述步骤i)中定容溶剂为叔丁基甲醚、水、乙腈、乙酸乙酯、二氯甲烷、丙酮、甲醇、甲醇-水的混合溶剂(V/V=75/25)、乙腈-0.3%乙酸水溶液(V/V=65/35)、乙腈-0.3%乙酸水溶液(V/V=55/45)、乙腈-0.3%乙酸水溶液(V/V=75/25)或乙腈-水的混合溶剂(V/V=75/25)。
进一步地,所述乙腈和0.3%乙酸水溶液的混合溶液为乙腈-0.3%乙酸水溶液(V/V=65/35)、乙腈-0.3%乙酸水溶液(V/V=55/45)或乙腈-0.3%乙酸水溶液(V/V=75/25)。
进一步地,所述过滤采用脱脂棉过滤。
进一步地,所述有机滤膜是孔径为0.45μm或0.22μm的聚四氟乙烯有机滤膜。
进一步地,所述5种松香酸为枞酸、新枞酸、左旋海松酸、海松酸和脱氢松香酸。
进一步地,所述浓缩为真空减压浓缩或氮气吹干浓缩。
本发明的有益效果
本发明分别研究了鞋材用胶黏剂、纸张、松香皂和油墨等4类产品中5种松香酸的检测方法,该方法采用超声波辅助溶剂对样品进行萃取,经萃取液浓缩后进行HPLC-MS/MS检测,所建方法具有良好的重复性和回收率,检测限低于50μg/kg,能有效满足各类产品中5种松香酸的测试要求。
在使用waters T3色谱柱分离5种松香酸时,液相色谱的最优化条件为:流动相为乙腈:水=65:35,流速为0.3mL/min,质谱检测的定量离子对分别为:m/z 303.2/257.6(枞酸);m/z 303.2/257.6(新枞酸);m/z 303.2/257.6(左旋海松酸);m/z 303.2/257.6(海松酸);m/z 301.2/172.9(脱氢松香酸)。
在20ug/L至1000.0ug/L范围内,5种松香酸的线性相关系数均大于0.995;胶黏剂、纸张、松香皂、油墨等4类物质中5种松香酸的测定低限低至50ug/kg,4类产品方法回收率在86%~109%,相对标准偏差(RSD)在8.0%以内,表明方法具有良好的重复性和可靠性。
本发明提供的操作方法简单,可操作性强,精密度高,重现性好,有效保证数据的准确性,检测限低,成本低,适合工业应用。
附图说明
图1是不同流动相比例下各种松香酸的分离图;
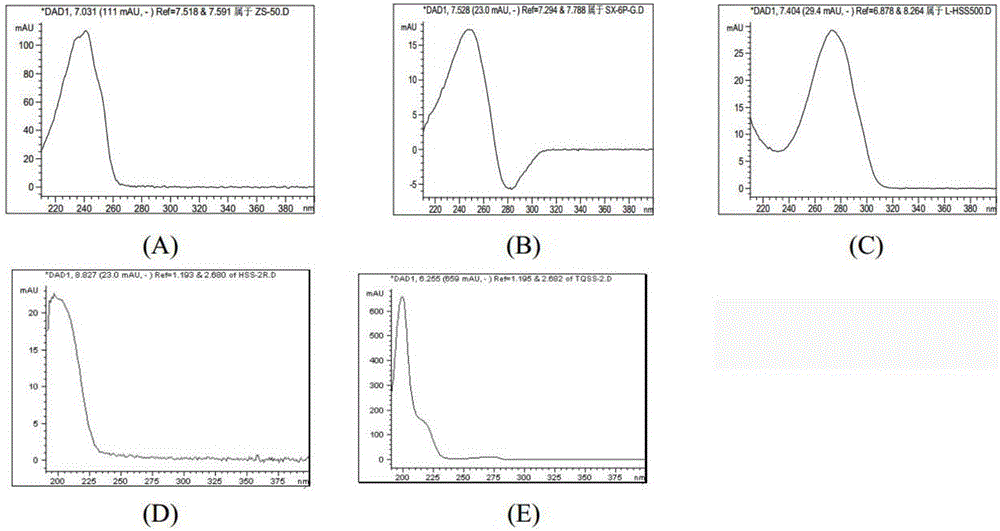
图2是5种松香酸的光谱图;
图3是正模式下枞酸和脱氢枞酸的总离子流图和质谱图。
图中,(A)枞酸,(B)新枞酸,(C)左旋海松酸,(D)海松酸,(E)脱氢松香酸。
具体实施方式
仪器与试剂
枞酸,纯度98.0%,美国Chroma Dex公司;新枞酸,纯度99.0%,加拿大Sigma公司;海松酸,纯度≧98.0%,加拿大Sigma公司;左旋海松酸,纯度≧98.0%,加拿大Sigma公司;脱氢松香酸,纯度≧98.0%,加拿大Toronto Research Chemicals公司。
乙腈、甲醇、叔丁基甲醚、乙酸乙酯、二氯甲烷、丙酮均为色谱纯,美国M-TEDIA公司;水为超纯水。
Agilent 6460B高效液相色谱质谱/质谱联用仪,美国安捷伦公司;
KQ-500E型数控超声波清洗器,输出功率:420W,频率:40kHz,昆山超声仪器公司;
聚四氟乙烯有机系滤膜,孔径0.22μm;
聚四氟乙烯有机系滤膜,孔径0.22μm,上海兴业净化材料厂;
管状硬质玻璃提取器,60mL,配聚四氟乙烯旋盖,德国CNW科技公司。
为使本领域技术人员更好地理解本发明的技术方案,下面结合附图和具体实施方式对本发明作进一步描述。
检测条件的优化
1、样品提取条件的优化
为减少操作步骤,获得尽可能高的提取效率,采用超声波辅助溶剂提取样品。针对胶黏剂、纸张、松香皂和油墨等4中产品的提取溶剂进行了考察,分别考察了乙酸乙酯、叔丁基甲醚、二氯甲烷、丙酮、甲醇、乙腈、甲醇-水的混合溶剂(V/V=75/25)、乙腈-0.3%乙酸水溶液的混合溶剂的提取效果,比较五种溶剂对样品的提取效率。在综合考虑提取效率、提取溶剂毒性和提取溶剂的沸点的前提下,胶黏剂中以叔丁基甲醚为最佳提取溶剂;纸张以甲醇-水的混合溶剂(V/V=75/25)为最佳提取溶剂;松香皂以乙腈-0.3%乙酸水溶液(V/V=65/35)为最佳提取溶剂;油墨以乙酸乙酯为最佳提取溶剂。
2、色谱条件的优化
(1)色谱柱的选择:分别考察了Eclipse Plus C18,2.1mm×50mm×1.8μm和Waters Atlantis T3,100×2.1mm,3μm两种类型的色谱柱对分离效果。通过实验发现采用Waters Atlantis T3,100×2.1mm,3μm,对目标物质的保留效果好,可将各松香酸分离,取得理想的分离效果。
(2)流动相及洗脱条件的选择:
不同的流动相对目标物质的分离效果及色谱峰形通常有显著影响。分别采用甲醇-水、乙腈-水体系进行条件的选择,实验结果表明,当流动相采用甲醇-水或者乙腈-水作为流动相时,5种目标物质均无法出峰,即目标物质无法形成稳定的正离子模式。当在流动相中加入一定的乙酸,5种目标物质出现色谱图,随着乙酸浓度的提高,目标物质的峰面积增加,当乙酸浓度达到0.3%时后,目标物质的峰面积趋于平缓。太低的pH值会影响色谱柱寿命,因此选择乙酸浓度为0.3%。图1为乙腈-0.3%乙酸水溶液比例分别为55:45、65:35和75:25时各种松香酸的分离情况。流动相比例为55:45时,样品出峰时间长,峰型特别宽,海松酸和枞酸完全重叠,达不到分析目的;当流动相比例为64:36到68:32之间时,目标物质出峰时间也还是较长,但是脱氢松香酸、新枞酸、左旋海松酸能完成分离,海松酸和枞酸能部分分离,可以进行定量分析;当流动相比例大等于68:32时,目标物质出峰时间变短,海松酸和枞酸完全重叠,变成一个峰,无法分析。因此选择流动相为乙腈-0.3%乙酸水溶液的混合溶剂(V/V=65/35)。
流速影响峰面积:考察流速对目标物质分离情况的影响。当流动相流速小于0.25mL/min时,目标物质出峰时间长,峰型宽;当流动相流速大于0.4mL/min时,海松酸和新枞酸完全重叠,脱氢松香酸、新枞酸峰面积变小,可能是流速过大,影响了母离子的离子化效率。因此流速选择0.3mL/min。
(3)吸收峰的选择:具体如图2所示,图2为5种松香酸的光谱图。光谱图显示,枞酸最大吸收波长在241nm处,在265nm以上和200nm以下几乎无吸收峰;新枞酸最大吸收波长在254nm处,在280nm以上和200nm以下几乎无吸收峰;左旋海松酸最大吸收波长在274nm,在240nm以下吸收峰较小。海松酸和脱氢松香酸最大吸收波长为200左右nm,在240nm以上几乎无吸收峰。
由于这些物质为同分异构体,相互之间无法完全分离,因此本发明在后续的实施例中在吸收波长选择中没有选择各种物质的最佳吸收波长:枞酸和新枞酸选择240nm,在此波长处可以避免左旋海松酸和异海松酸的干扰,而左旋海松酸选择280nm,在此波长处可以避免新枞酸和枞酸的干扰,而海松酸和脱氢松香酸选择200nm,在此波长处可以避免枞酸、新枞酸和海松酸的干扰。
3、质谱测定条件的优化
由于样品种类繁多,为保证分析测定的准确性,选择采用液相色谱‐串联质谱(LC‐MS/MS)对松香酸进行测定。分别考察了电喷雾正、负两种离子化方式,实验显示,ESI+模式下,枞酸、新枞酸、左旋海松酸、海松酸均出现303.1的准分子离子峰,脱氢松香酸出现301.4的准分子离子峰。采用Product Ion SIM模式对5种松香酸进行研究。图3中(a)和(b)分别为枞酸和脱氢松香酸的总离子流图,图3(c)和(d)分别为枞酸和脱氢松香酸的质谱图。从图3中可见,枞酸的碎片离子可以选择257.6,123.1,148.9等;脱氢松香酸的碎片离子可以选择172.9,240.0等选择合适的毛细管出口电压(Fragmentor)和碰撞能量(collision energy),可以找到传输效率最佳的母离子和响应最好的选择离子。
实施例1胶黏剂中松香酸的检测
准确称取309胶黏剂样品1g,置于管状硬质玻璃提取器中,加入10mL叔丁基甲醚,在60℃下进行第一次超声提取30min,过滤;样品中再次加入3mL叔丁基甲醚,在60℃下进行第二次超声提取30min,过滤;将两次过滤的滤液合并后进行浓缩,加入乙腈-0.3%乙酸水溶液的混合溶剂(V/V=65/35)在25mL容量瓶中进行定容,取样品溶液经0.45μm有机滤膜过滤后,用HPLC-MS进行测定,选择两级质谱的特定离子对,按色谱峰的保留时间定性,以峰面积外标法定量;
液相色谱条件:
液相色谱柱:Waters Atlantis T3,100×2.1mm,3μm;
流动相:乙腈:0.3%乙酸水溶液(V/V)=65:35,流量为0.3mL·min-1;
进样量为5uL;波长为200nm、240nm、280nm,柱温40℃;
质谱条件为:
a)离子源:电喷雾离子源(ESI+);
b)离子模式:正离子模式
c)检测方式:多反应离子监测(MRM);
d)定性离子对:m/z 303.2/148.9和m/z 303.2/123.0(枞酸);m/z 303.2/148.9和m/z303.2/123.0(新枞酸);m/z 303.2/148.9和m/z 303.2/123.0(左旋海松酸);m/z 303.2/148.9和m/z 303.2/123.0(海松酸);m/z 301.2/172.9和m/z 301.2/240.0(脱氢松香酸);
j)定量离子对:m/z 303.2/257.6(枞酸);m/z 303.2/257.6(新枞酸);m/z 303.2/257.6(左旋海松酸);m/z 303.2/257.6(海松酸);m/z 301.2/172.9(脱氢松香酸);
k)干燥气温度:325℃;
l)干燥气流速:10L/min;
m)雾化气压力:40Psi;
n)电喷雾电压:4000V;
o)离子源温度:100℃;
p)毛细管出口电压和碰撞电压如下表1:
表1
具体检测结果如表2所示。
实施例2纸张中松香酸的检测
将纸张进行剪碎成约1mm×1mm的小碎片,准确称取纸张样品1g,置于管状硬质玻璃提取器中,加入10mL甲醇-水的混合溶剂(V/V=75/25),在60℃下进行第一次超声提取30min,过滤;样品中再次加入10mL甲醇-水的混合溶剂(V/V=75/25),在60℃下进行第二次超声提取30min,过滤;将两次过滤的滤液合并后进行浓缩,加入乙腈-0.3%乙酸水溶液的混合溶剂(V/V=65/35)溶解后在25mL容量瓶中进行定容,取样品溶液经0.45μm有机滤膜过滤后,用HPLC-MS进行测定,选择两级质谱的特定离子对,按色谱峰的保留时间定性,以峰面积外标法定量;
液相色谱条件:
液相色谱柱:Waters Atlantis T3,100×2.1mm,3μm;
流动相:乙腈:0.3%乙酸水溶液(V/V)=65:35,流量为0.3mL·min-1;
进样量为5uL;波长为200nm、240nm、280nm,柱温40℃;
质谱条件为:
a)离子源:电喷雾离子源(ESI+);
b)离子模式:正离子模式
c)检测方式:多反应离子监测(MRM);
d)定性离子对:m/z 303.2/148.9和m/z 303.2/123.0(枞酸);m/z 303.2/148.9和m/z303.2/123.0(新枞酸);m/z 303.2/148.9和m/z 303.2/123.0(左旋海松酸);m/z 303.2/148.9和m/z 303.2/123.0(海松酸);m/z 301.2/172.9和m/z 301.2/240.0(脱氢松香酸);
j)定量离子对:m/z 303.2/257.6(枞酸);m/z 303.2/257.6(新枞酸);m/z 303.2/257.6(左旋海松酸);m/z 303.2/257.6(海松酸);m/z 301.2/172.9(脱氢松香酸);
k)干燥气温度:325℃;
l)干燥气流速:10L/min;
m)雾化气压力:40Psi;
n)电喷雾电压:4000V;
o)离子源温度:100℃;
p)毛细管出口电压和碰撞电压如下表1;
具体检测结果如表2所示。
实施例3松香皂中松香酸的检测
将松香皂进行剪碎成约1mm×1mm的小碎片,准确称取样品1g,置于管状硬质玻璃提取器中,加入10mL乙腈-0.3%乙酸水溶液(V/V=65/35),在60℃下进行第一次超声提取30min,过滤;样品中再次加入10mL乙腈-0.3%乙酸水溶液(V/V=65/35),在60℃下进行第二次超声提取30min,过滤;将两次过滤的滤液合并后加入乙腈-0.3%乙酸水溶液的混合溶剂(V/V=65/35)在容量瓶中进行定容为25mL,取样品溶液经0.45μm有机滤膜过滤后,用HPLC-MS进行测定,选择两级质谱的特定离子对,按色谱峰的保留时间定性,以峰面积外标法定量;
液相色谱条件:
液相色谱柱:Waters Atlantis T3,100×2.1mm,3μm;
流动相:乙腈:0.3%乙酸水溶液(V/V)=65:35,流量为0.3mL·min-1;
进样量为5uL;波长为200nm、240nm、280nm,柱温40℃;
质谱条件为:
a)离子源:电喷雾离子源(ESI+);
b)离子模式:正离子模式
c)检测方式:多反应离子监测(MRM);
d)定性离子对:m/z 303.2/148.9和m/z 303.2/123.0(枞酸);m/z 303.2/148.9和m/z303.2/123.0(新枞酸);m/z 303.2/148.9和m/z 303.2/123.0(左旋海松酸);m/z 303.2/148.9和m/z 303.2/123.0(海松酸);m/z 301.2/172.9和m/z 301.2/240.0(脱氢松香酸);
j)定量离子对:m/z 303.2/257.6(枞酸);m/z 303.2/257.6(新枞酸);m/z 303.2/257.6(左旋海松酸);m/z 303.2/257.6(海松酸);m/z 301.2/172.9(脱氢松香酸);
k)干燥气温度:325℃;
l)干燥气流速:10L/min;
m)雾化气压力:40Psi;
n)电喷雾电压:4000V;
o)离子源温度:100℃;
p)毛细管出口电压和碰撞电压如下表1;
具体检测结果如表2所示。
实施例4油墨中松香酸的检测
将油墨材料在玻璃板上自然晾干后,剪碎;印刷制品用刮刀刮下,若无法刮下,则直接连基体材料一同处理,准确称取样品1g,置于管状硬质玻璃提取器中,加入10mL乙酸乙酯,在60℃下进行第一次超声提取30min,过滤;样品中再次加入10mL乙酸乙酯,在60℃下进行第二次超声提取30min,过滤;将两次过滤的滤液合并后进行浓缩,加入乙腈-0.3%乙酸水溶液的混合溶剂(V/V=65/35)溶解后在25mL容量瓶中定容,取样品溶液经0.45μm有机滤膜过滤后,用HPLC-MS进行测定,选择两级质谱的特定离子对,按色谱峰的保留时间定性,以峰面积外标法定量;
液相色谱条件:
液相色谱柱:Waters Atlantis T3,100×2.1mm,3μm;
流动相:乙腈:0.3%乙酸水溶液(V/V)=65:35,流量为0.3mL·min-1;
进样量为5uL;波长为200nm、240nm、280nm,柱温40℃;
质谱条件为:
a)离子源:电喷雾离子源(ESI+);
b)离子模式:正离子模式
c)检测方式:多反应离子监测(MRM);
d)定性离子对:m/z 303.2/148.9和m/z 303.2/123.0(枞酸);m/z 303.2/148.9和m/z303.2/123.0(新枞酸);m/z 303.2/148.9和m/z 303.2/123.0(左旋海松酸);m/z 303.2/148.9和m/z 303.2/123.0(海松酸);m/z 301.2/172.9和m/z 301.2/240.0(脱氢松香酸);
j)定量离子对:m/z 303.2/257.6(枞酸);m/z 303.2/257.6(新枞酸);m/z 303.2/257.6(左旋海松酸);m/z 303.2/257.6(海松酸);m/z 301.2/172.9(脱氢松香酸);
k)干燥气温度:325℃;
l)干燥气流速:10L/min;
m)雾化气压力:40Psi;
n)电喷雾电压:4000V;
o)离子源温度:100℃;
p)毛细管出口电压和碰撞电压如下表1;
具体结果如表2所示:
表2不同样品中松香酸的含量(mg/kg)
实施例5标准线性方程及检出限
5种松香酸标准储备液的配制:称取适量的5种松香酸,用乙腈配成100ug/mL的标准储备液,使用乙腈-0.3%乙酸水溶液的混合溶剂(V/V=65/35)逐级稀释成浓度为20.0ug/L,50.0ug/L,100.0ug/L,500.0ug/L,1000.0ug/L的标准工作液,供液相色谱串联质谱分析测定,绘制标准工作曲线,在20.0~1000.0ug/L的浓度范围内,相关系数均大于0.999,具体如表3所示。
于胶黏剂、纸张、松香皂、油墨等4种空白基质中加入低浓度的标准溶液,按照本发明实施例1~4所述处理方式进行处理分析后,测试,采用Agilent6460自带软件的信噪比工具计算信噪比S/N,检出限按照3倍信噪比计算,定量检出限按照10倍信噪比计算,结果见表3。从表3中结果可知,在考虑基体干扰情况下,可将5种物质脱氢松香酸、枞酸和新松酸定量检出限设为50ug/kg。
表3方法的线性关系和检出限
实施例6回收和精密度试验
选用胶黏剂、纸张、松香皂、油墨等4类产品空白样品为基质,加入浓度为20.0ug/L、100.0ug/L、1000.0ug/L三个不同浓度的5种松香酸的混合溶液,按本发明实施例1~4所述处理方式进行处理分析后,平行测定5次,测回收率和精密度,结果见表4,从表4中结果可知,4类产品方法回收率在86%~109%,相对标准偏差(RSD)在8.0%以内,表明方法具有良好的重复性和可靠性。
表4方法的回收率和精密度(n=5)
可以理解的是,以上实施方式仅仅是为了说明本发明的原理而采用的示例性实施方式,然而本发明并不局限于此。对于本领域内的普通技术人员而言,在不脱离本发明的精神和实质的情况下,可以做出各种变型和改进,这些变型和改进也视为被包含在本发明的保护范围内。
- 还没有人留言评论。精彩留言会获得点赞!
- 利用超高效液相色谱‑串联质谱法同时检测水果中3种增甜剂的方法与流程
- 利用超高效液相色谱‑串联质谱法同时检测果汁中7种甜味剂的方法与流程
- 采用反相高效液相色谱法分离手性对映体的方法及羟丙基‑β‑环糊精的应用与流程
- 利用反相高效液相色谱法测定桦木酮酸‑氨基酸衍生物含量的方法与流程
- 一种超高效液相色谱串联质谱的检测方法与流程
- 免疫亲和柱净化‑超高效液相色谱‑串联质谱检测鱼虾中3‑甲基‑喹噁啉‑2‑羧酸的方法与流程
- 高效液相色谱串联质谱检测血清中五种甘氨结合型胆汁酸的方法与流程
- 一种超高效液相质谱联用测定吴茱萸配方颗粒中10种成分的定量方法与流程
- 一种固相萃取‑高效液相色谱‑串联质谱法检测茶叶中高氯酸盐的方法与流程
- 一种反相液相色谱法检测雷替曲塞对映异构体的方法与流程