预测氨基酸突变的方法及系统与流程
本发明涉及生物信息技术领域,尤其涉及一种预测氨基酸突变的方法及系统。
背景技术:
氨基酸突变也被称为非同义的单核苷酸突变,是关于人类疾病变种研究中价值最大的一部分。氨基酸突变是由于一些单个碱基的改变,造成蛋白质产物中氨基酸序列的改变。氨基酸的改变会影响蛋白质的稳定性、相互作用和酶的活性,从而导致疾病的发生。根据最新的全人类基因组测序的结果表明,每个人都会有三到五百万个氨基酸突变,而且这一数据仍然在快速的增长。在众多的氨基酸突变中,一些突变会导致疾病的发生,其他的则是对蛋白质功能没有影响的中性突变。随着单核苷酸多态性等位基因分型和下一代DNA测序技术等基因组分析技术的快速发展,产生了大量关于氨基酸突变的数据。这些数据通过研究发现,绝大多数的氨基酸突变都是疾病形成的潜在因素,但是通过生物实验来测定大量突变的表现型和生物化学性质比较耗时耗力,且成本花费大。然而,如果采用数据挖掘的方法对大量突变数据进行预测,不仅预测精度高、方便、快速,而且节约成本。
目前已经有许多计算方法运用于氨基酸突变的预测。这些方法主要使用的是统计原理和机器学习算法进行预测分类。例如:SIFT、SNAP、PolyPhen2、FunSAV和SusPect等方法。它们的输入特征包括氨基酸序列、3D结构、氨基酸的理化性质、进化信息和残基互联网络等特征。而且许多方法已经被做成独立软件或者网站服务器,可供研究团体免费使用。但现有的方法预测精度不高,所以还有很大的提升空间。因此,如果能运用一种有效的方法,把有害的和中性的氨基酸突变相互区分开,将更好的理解基因型和表现型的相互关系,进而揭开基因遗传突变致病的内部细节。
技术实现要素:
本发明目的在于公开一种预测氨基酸突变的方法及系统,以提高预测的精度和效果,有效解决生物实验的盲目性和成本高等问题。
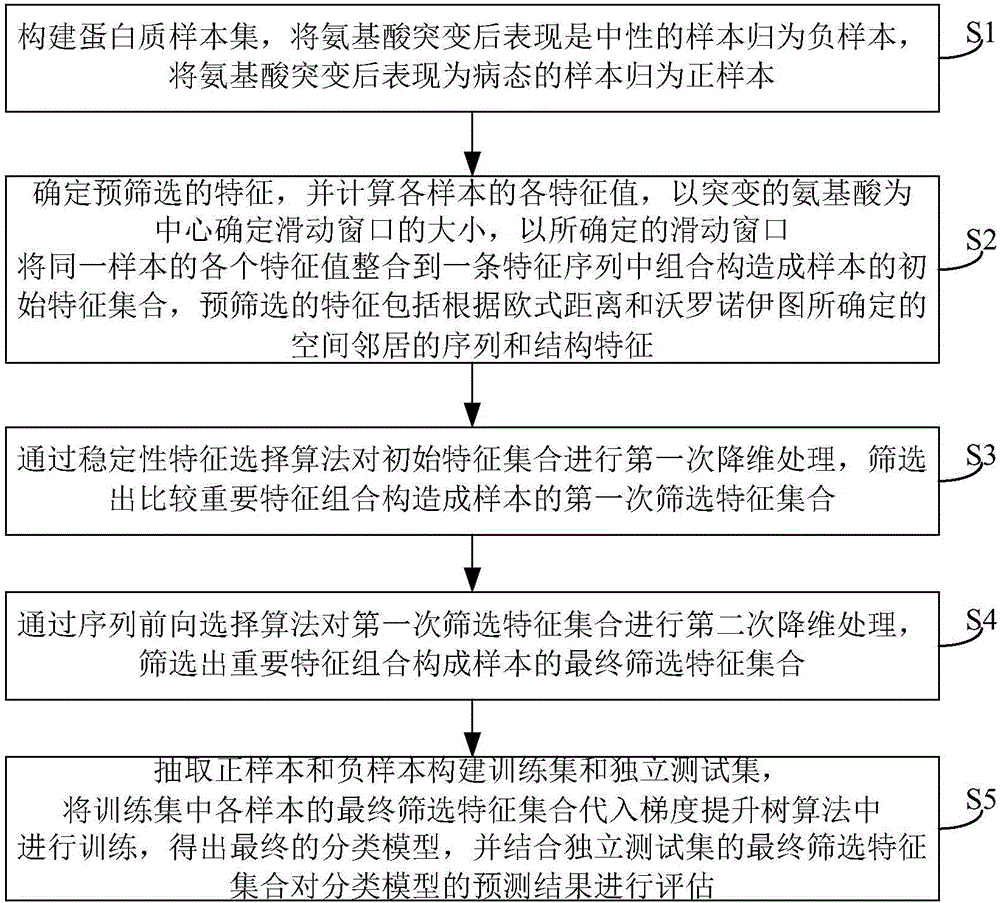
为实现上述目的,本发明公开了一种预测氨基酸突变的方法,包括:
构建蛋白质样本集,将氨基酸突变后表现是中性的样本归为负样本,将氨基酸突变后表现为病态的样本归为正样本;
确定预筛选的特征,并计算各样本的各特征值,以突变的氨基酸为中心确定滑动窗口的大小,以所确定的滑动窗口将同一样本的各个特征值整合到一条特征序列中组合构造成样本的初始特征集合,所述预筛选的特征包括根据欧式距离和沃罗诺伊图所确定的空间邻居的序列和结构特征;
通过稳定性特征选择算法对所述初始特征集合进行第一次降维处理,筛选出比较重要特征组合构造成样本的第一次筛选特征集合;
通过序列前向选择算法对所述第一次筛选特征集合进行第二次降维处理,筛选出重要特征组合构成样本的最终筛选特征集合;
抽取正样本和负样本构建训练集和独立测试集,将所述训练集中各样本的最终筛选特征集合代入梯度提升树算法中进行训练,得出最终的分类模型,并结合所述独立测试集的最终筛选特征集合对所述分类模型的预测结果进行评估。
为实现上述目的,本发明还公开了一种预测氨基酸突变的系统,包括:
第一处理模块,用于构建蛋白质样本集,将氨基酸突变后表现是中性的样本归为负样本,将氨基酸突变后表现为病态的样本归为正样本;
第二处理模块,用于确定预筛选的特征,并计算各样本的各特征值,以突变的氨基酸为中心确定滑动窗口的大小,以所确定的滑动窗口将同一样本的各个特征值整合到一条特征序列中组合构造成样本的初始特征集合,所述预筛选的特征包括根据欧式距离和沃罗诺伊图所确定的空间邻居的序列和结构特征;
第三处理模块,用于通过稳定性特征选择算法对所述初始特征集合进行第一次降维处理,筛选出比较重要特征组合构造成样本的第一次筛选特征集合;
第四处理模块,用于通过序列前向选择算法对所述第一次筛选特征集合进行第二次降维处理,筛选出重要特征组合构成样本的最终筛选特征集合;
第五处理模块,用于抽取正样本和负样本构建训练集和独立测试集,将所述训练集中各样本的最终筛选特征集合代入梯度提升树算法中进行训练,得出最终的分类模型,并结合所述独立测试集的最终筛选特征集合对所述分类模型的预测结果进行评估。
本发明具有以下有益效果:
通过提取多种氨基酸特征进行两次降维处理得出重要特征,并基于降维后的重要特征进行分类模型的构建及评估,提高了预测的精度和效果,有效解决生物实验的盲目性和成本高等问题。
下面将参照附图,对本发明作进一步详细的说明。
附图说明
构成本申请的一部分的附图用来提供对本发明的进一步理解,本发明的示意性实施例及其说明用于解释本发明,并不构成对本发明的不当限定。在附图中:
图1是本发明优选实施例公开的预测氨基酸突变的方法流程图;
图2为5折验证下两步特征选择方法与其他方法性能比较的ROC曲线;
图3为梯度提升树算法与其他前人用到的分类方法性能比较;
图4为5折验证下本发明方法与其他方法在训练集上性能比较的ROC曲线;
图5为5折验证下本发明方法与其他方法在独立测试集上性能比较的ROC曲线。
具体实施方式
以下结合附图对本发明的实施例进行详细说明,但是本发明可以由权利要求限定和覆盖的多种不同方式实施。
实施例1
本实施例公开一种预测氨基酸突变的方法,如图1所示,包括:
步骤S1、构建蛋白质样本集,将氨基酸突变后表现是中性的样本归为负样本,将氨基酸突变后表现为病态的样本归为正样本。
该步骤中,负样本的数据可从Ensemble human variation数据库中提取。正样本的数据可从UniProt human sequence variations数据库中提取。
较佳的,在构建蛋白质样本集的过程中,可以先对蛋白质序列相似性大于0.4的重复性数据做剔除处理。
步骤S2、确定预筛选的特征,并计算各样本的各特征值,以突变的氨基酸为中心确定滑动窗口的大小,以所确定的滑动窗口将同一样本的各个特征值整合到一条特征序列中组合构造成样本的初始特征集合,预筛选的特征包括根据欧式距离和沃罗诺伊图所确定的空间邻居的序列和结构特征。在该步骤中,以突变的氨基酸为中心,每个特征的计算都与其左右相邻的若干残基的特征值进行关联,例如滑动窗口为21,则所择取的左右相邻的残基数量各为10。而且通常,每个特征值的计算是单独实现的,即各个特征值的具体计算算法不一样,从而需要将同一样本的各个特征值整合到一条特征序列中。
预筛选的特征还包括但不限于:物理化学特征,特定位置得分矩阵,溶剂可及性表面积,螺旋转角,替换矩阵,二级结构,保守性分数,残基结构熵值及残基相互联系的网络特征等等。
在该步骤中,可以通过三维坐标计算,根据欧式距离(Euclidean distance)和沃罗诺伊图(Voronoi diagram)确定蛋白质样本中突变氨基酸所处链式三维空间中的邻居残基,进而考虑空间邻居的序列和结构特征,而不是仅仅只考虑氨基酸序列相邻的邻居特征;从而扩展了特征的纳入范畴,而且在大量的实验中,该空间邻居的序列和结构特征在后续的降维过程中都得以保留,并对最终预测结果产生了实质性的影响,从而使得该考虑相比现有的特征纳入范畴具有显著的进步。
步骤S3、通过稳定性特征选择算法对初始特征集合进行第一次降维处理,筛选出比较重要特征组合构造成样本的第一次筛选特征集合。
在该步骤中,稳定性特征选择(Stability feature selection)方法是一个较新颖的特征选择方法。它是在各个不同的数据子集上使用特征选择算法,不同的数据子集包含不同的特征。这个过程会重复若干次后,选择结果会被汇总,通过从子集中查找一个特征有多少次被选为重要特征,被选的次数越多的特征越重要。其中重要特征的分数会接近1,因为他们总是被选中。差一点的特征会是介于1到0之间,因为他们也会在子集中被选中。而最没有用的特征分数会接近0,因为他们从来没有被选中。分数就代表了特征的重要性,越重要的特征就越能准确分类对数据。
步骤S4、通过序列前向选择算法对第一次筛选特征集合进行第二次降维处理,筛选出重要特征组合构成样本的最终筛选特征集合。
在该步骤中,序列前向选择(Sequential forward selection)算法是一种简单的贪心算法,每次都选择一个使得评价函数的取值达到最优的特征加入,并在加入新特征导致评价函数取值变劣的临界前终止。
在本实施例中,采用上述稳定性特征选择算法和序列前向选择算法还具有一个突出的特点,其在筛选的过程中,由于不对特征及相应的特征值进行相关的数据转换,从而可以观测最终保留的特征及相应的特征值,藉此,该步骤使得本实施例能在不断的实验过程中,可进一步为扩展上述特征的纳入范畴提供相应的参考。
为便于描述,上述步骤S3及步骤S4的特征筛选组合简称为“两步特征选择方法”
步骤S5、抽取正样本和负样本构建训练集和独立测试集,将训练集中各样本的最终筛选特征集合代入梯度提升树算法中进行训练,得出最终的分类模型,并结合独立测试集的最终筛选特征集合对分类模型的预测结果进行评估。其中,对于分类模型的构建及评估,本领域技术人员所熟知的,大多由交叉验证阶段(例如:5折交叉验证)和独立测试阶段组成,并可用现有评估标准:准确度(ACC),Matthews相关系数(MCC),真阴性率(Sp),灵敏度(Sn),ROC曲线以及ROC曲线下面积(AUC)等来对预测结果进行评估。藉此,当用户通过本实施例所确定的分类模型(即预测模型)来进行预测时,只需要输入相关蛋白质的名称,氨基酸名称及所在链中的位置(必要时,还可加入相应的最终筛选特征集合中的相应特征)等信息,即可预测出该氨基酸突变结果的概率值,例如,呈中性的概率值或呈病态的概率值。
在该步骤中,梯度提升树算法(Gradient tree boosting algorithm,简称GTB)通过构建多个子分类器,并且每个子分类器都是在前一个的基础上进行建立的。最后每个子分类器对氨基酸突变的性状类别进行投票,然后选择票数最多的类别作为最终预测类别。本实施例可选的梯度提升树算法如下:
输入:
数据集:x是样本的特征向量,y是类标(正样本或负样本)。
损失函数:L(y,Θ(x));y是真实的类标(-1代表负样本),Θ(x)是决策函数。
迭代次数=M;M代表迭代多少次,即构建多少个分类模型。
输出:
1)、初始化(初始化决策函数,N是训练集中氨基酸的个数,即样本个数;yi是类标;c就是当前要求的分类器模型(c是classify的简写))。
2)、从m=1到M,重复步骤3)到6)。
3)、计算损失函数的负梯度作为残差值。
4)、通过使用输入x的损失函数找到一个合适的分类模型,并得到βmh(x;αm)的估计αm;βm和αm分别是第m个分类树的权重和参数向量,h(x;αm)是第m个分类树。
5)、通过最小化L(yi,Θm-1(xi)+βh(xi,αm))得到估计βm,最小化损失函数。
6)、更新Θm(x)=Θm-1(x)+βmh(x;αm);Θm-1(x)是前一个决策函数,Θm(x)是目前所求的决策函数,通过迭代的计算,使用前一步的结果来计算后面的。
7)、返回用最终的决策函数作为梯度提升树的分类模型,因为最终的损失函数值最小,即真实值和预测值之间的误差最小。为最终的得到的梯度提升树模型。
在对比实验中,可从上述数据库得到670个蛋白质结构数据,其中包含963个中性突变和1006疾病相关突变;从中随机的选出816疾病相关突变和776个中性突变作为训练集数据,剩下190个疾病相关突变和187个中性突变作为独立测试集。通过实现5折交叉验证来对本发明进行性能评估。在5折交叉验证中,我们在训练集上将两步特征选择方法(two-step)和其他三种当今通用的特征选择算法(RF:随机森林算法,RFE:基于SVM的递归特征消除算法,mRMR:最小冗余最大相关算法)进行了比较,通过图2给出的ROC曲线可以看出,本发明的两步特征选择方法所得出的AUC(ROC曲线的面积)值最高,这表明两步特征选择算法优于其他方法。接下来我们用梯度梯度提升树算法(GTB)和前人用过的随机森林(RF)算法和支持向量机(SVM)进行了比较,通过图3的柱状图,我们看出梯度提升树算法预测效果更高。最后本实施例所公开的预测方法(PredSAV)与其它目前最先进的方法(包括:FunSAV、PolyPhen2、SusPect、SIFT、SNAP等)基于同样的训练集及独立测试样本集进行了比较,如图4和图5所示,可以看出本发明的方法比其他方法预测性能更好。
值得说明的是,本实施中的上述稳定性特征选择算法、序列前向选择算法及梯度提升树算法是国内外在其他应用领域都比较常规的算法,其涉及多方面内容且不是本发明首创,故本发明仅对其实现机理进行简述,不一一展开详述。
综上,本实施例公开的预测氨基酸突变的方法,通过提取多种氨基酸特征进行两次降维处理得出重要特征,可以从初始预筛选的上千维特征中筛选出几十维最为重要的特征,并基于降维后的重要特征进行分类模型的构建及评估,提高了预测的精度和效果,有效解决生物实验的盲目性和成本高等问题。
实施例2
与上述方法实施例相对应的,本实施例公开一种预测氨基酸突变的系统,包括下述的第一至第五处理模块。各模块的功能分述如下:
第一处理模块,用于构建蛋白质样本集,将氨基酸突变后表现是中性的样本归为负样本,将氨基酸突变后表现为病态的样本归为正样本。其中,负样本的数据可从Ensemble humanvariation数据库中提取;正样本的数据可从UniProt human sequence variations数据库中提取。较佳的,该第一处理模块还用于在构建蛋白质样本集的过程中,对蛋白质序列相似性大于0.4的重复性数据做剔除处理,进而提高数据样本的含金量。
第二处理模块,用于确定预筛选的特征,并计算各样本的各特征值,以突变的氨基酸为中心确定滑动窗口的大小,以所确定的滑动窗口将同一样本的各个特征值整合到一条特征序列中组合构造成样本的初始特征集合,预筛选的特征包括根据欧式距离和沃罗诺伊图所确定的空间邻居的序列和结构特征。可选的,该第二处理模块所确定的预筛选的其他特征还包括:物理化学特征,特定位置得分矩阵,溶剂可及性表面积,螺旋转角,替换矩阵,二级结构,保守性分数,残基相互联系的网络特征及二进制编码等等。
第三处理模块,用于通过稳定性特征选择算法对初始特征集合进行第一次降维处理,筛选出比较重要特征组合构造成样本的第一次筛选特征集合。
第四处理模块,用于通过序列前向选择算法对第一次筛选特征集合进行第二次降维处理,筛选出重要特征组合构成样本的最终筛选特征集合。
第五处理模块,用于抽取正样本和负样本构建训练集和独立测试集,将训练集中各样本的最终筛选特征集合代入梯度提升树算法中进行训练,得出最终的分类模型,并结合独立测试集的最终筛选特征集合对分类模型的预测结果进行评估。
同理,本实施例公开的预测氨基酸突变的系统,通过提取多种氨基酸特征进行两次降维处理得出重要特征,可以从初始预筛选的上千维特征中筛选出几十维最为重要的特征,并基于降维后的重要特征进行分类模型的构建及评估,提高了预测的精度和效果,有效解决生物实验的盲目性和成本高等问题。
以上所述仅为本发明的优选实施例而已,并不用于限制本发明,对于本领域的技术人员来说,本发明可以有各种更改和变化。凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!