一种用于治疗肿瘤疾病的药物的制作方法
本发明涉及一种杂环化合物,特别涉及一种氧氮杂唑酮衍生物,属于生物医药领域。
背景技术:
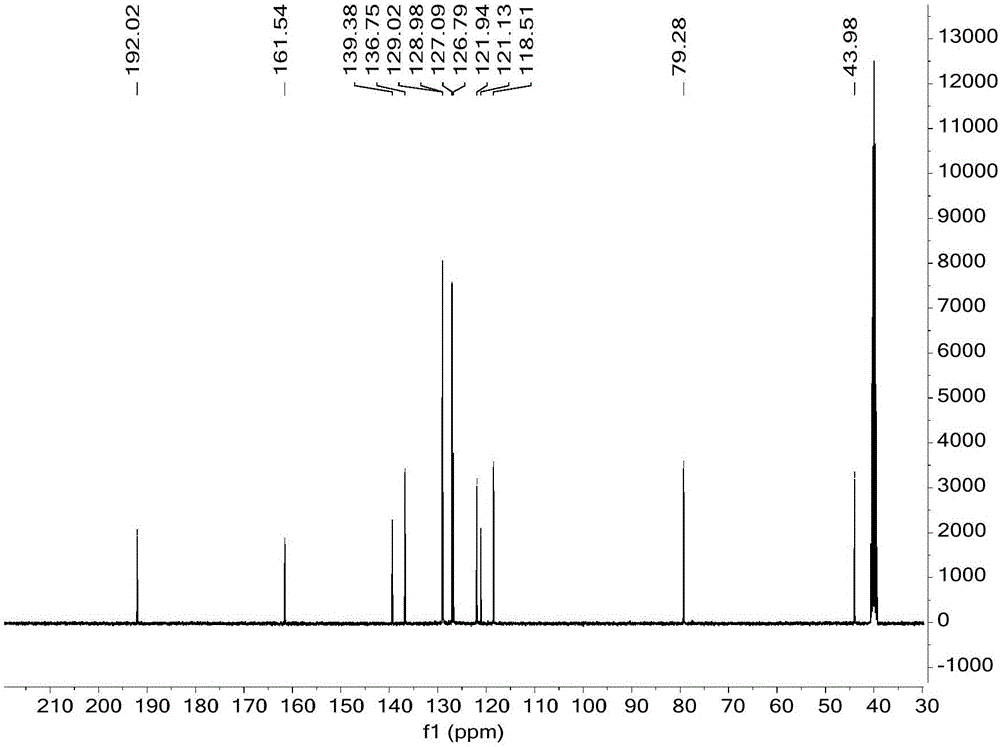
:现代社会生活节奏快,人们日常工作压力大,长期处于亚健康状态。自身免疫力的不足,导致各种外源性的病菌入侵和内源性的细胞异常生长比率持续增高,严重威胁人类生命。20世纪以来人类对于药物的研究逐渐转向有机化学合成,其中小分子化合物以其独特的空间立体结构、电子分布、活性基团的空间排布等特点,在各种疾病治疗中发挥出了突出的作用。氧氮杂唑酮衍生物是含氧原子,氮原子的杂环,它可以通过结构中的内酰胺与生物体内与肿瘤疾病相关的酶和受体2发生氢键作用。另一方面,结构中的芳香环与肿瘤疾病相关的酶和受体形成芳环堆叠作用。从而达到治疗肿瘤疾病的目的。技术实现要素:本发明的目的在于提供了一种具有式Ⅰ结构的氧氮杂唑酮衍生物,同时本发明还提供了制备该化合物的方法。本发明的另一目的是提供一种用于治疗肿瘤疾病的药物化合物或组合物,该药物化合物或组合物含有式Ⅰ氧氮杂唑酮衍生物、或其生物学可接受的盐,作为为活性成分。为了实现上述发明目的,本发明提供了以下技术方案:一种用于治疗肿瘤疾病的药物,该药物含有具有式Ⅰ所示结构式的氧氮杂唑酮衍生物或式Ⅰ化合物的生物学可接受的盐作为活性成分。其中,1)X是氧或硫;2)n=0-4;R1各自独立的选自:氢、卤素、羟基、氰基、硝基、氨基、C1-C7烷基、C1-C6烷氧基、C3-C7环烷基、C1-C6烷氧基羰基、C1-C6烷基羰基、氨基羰基、C1-C6烷基氨基羰基、取代氨基、恶唑基、噻唑基、哌啶基、六氢哌啶基、吡啶基、二氢吡啶基、四氢吡啶基、噻嗪基、吡咯基、咪唑基、吡唑基、嘧啶基、哌嗪基、吗啉基、取代哌嗪基、呋喃基、吡喃基、任选地被取代基Y取代的杂环基,任选地被取代基Y取代的芳基,任选地被取代基Y取代的杂芳基等;优选的,所述的杂环基是5-6个原子组成的杂环基团,杂环基上具有1-3个杂原子,其中的杂原子是氮、氧、硫中的至少一种。更优选的,所述的杂环是呋喃、吡啶、嘧啶、哌啶、噻吩、吡咯、噻唑、嘌呤、吡喃、噁唑、咪唑、吡嗪、喹啉等。上述的杂环基可以被1-3个低分子烃基取代,所述的低分子烃基是1-4个碳原子的烃基。所述的芳基是苯基、萘基、甲苯基、乙苯基、丙基苯基、乙烯基苯基、羟基苯基、二甲基苯基、甲乙基苯基、氯苯基、三氟甲基苯基、对氨基苯基、甲酸苯基等。其中所述取代基Y为氢原子、C1-C6烷基、卤代C1-C6烷基、卤素、羟基、硝基、芳基、氨基、C1-C6烷基氨基、C3-C7环烷基氨基、二(C1-C6烷基)氨基、氰基或C3-C7环烷基等;所述取代氨基包括苯基氨基、苯基甲基氨基、取代苯基氨基、甲基氨基、乙基氨基、丙基氨基、异丙基氨基、环丙基氨基、环己基氨基、哌啶基氨基、甲基哌啶基氨基、吡啶基氨基、哌嗪基氨基等;所述取代苯基氨基是指甲基苯基氨基、乙基苯基氨基、丙基苯基氨基、二甲基苯基氨基、甲基(乙基)苯基氨基、卤取代苯基氨基、硝基苯基氨基、苯基亚砜基苯基氨基、苯甲酮基苯基氨基和二苯甲酮基氨基中的一个;所述取代哌嗪基是指甲基哌嗪基、乙基哌嗪基、丙基哌嗪基、二甲基哌嗪基、甲基(乙基)哌嗪基、异丙基哌嗪基、卤取代哌嗪基;卤取代哌嗪基中卤素包括氟、氯、溴、碘。3)m=0-5;R2为各自独立的选自:氢、氟、氯、溴、羟基、氰基、硝基、C1-C4烷基、C1-C3烷氧基、C3-C7环烷基、卤代C1-C6烷基、C2-C6烯基、羟基取代的C1-C6烷基、二(C1-C6烷基)氨基-C1-C6烷基、氨基、C1-C6烷基氨基、C3-C7环烷基氨基、二(C1-C6烷基)氨基、氨基-C1-C6烷基氨基、C1-C6烷氧基C1-C6烷基氨基、C1-C6烷氧基羰基、C1-C6烷基氨基、二(C1-C6烷氧基-C1-C6烷基)氨基、氨基羰基、C1-C6烷基氨基羰基、二(C1-C6烷基)氨基羰基、C3-C7环烷基氨基羰基、C1-C6烷氧基、C3-C7环烷氧基、羟基-C1-C6烷氧基、卤代C1-C6烷氧基、氨基C1-C6烷基、氨基C1-C6烷氧基、C1-C6烷基砜、C2-C6烯基砜、C3-C7环烷基砜、C3-C7环烷基、卤代C3-C7环烷基、杂环氧基、哌啶基氨基、N-甲基哌啶-4-羰基、哌嗪-C1-C6烷基、吡咯甲酰胺基、N-甲基哌啶甲酰胺基、或杂环C1-C6烷基氧基等。任意的被取代基Z单取代或双取代的氨基。所述的杂环氧基中的杂环是呋喃、吡啶、嘧啶、哌啶、噻吩、吡咯、噻唑、嘌呤、吡喃、噁唑、咪唑、吡嗪、喹啉等。其中所述取代基Z为可以是各自独立的取代基,双取代时也可以是相互连接形成的环形结构。当其为各自独立的取代基时,Z各自独立的选自:氢、卤素、氰基、硝基、C1-C7烷基、C1-C6烷氧基、C3-C7环烷基、C1-C6烷氧基羰基、C1-C6烷基羰基、氨基羰基,C1-C6烷基氨基羰基、芳香基羰基、任意地被取代基Y芳香基羰基、恶唑基、噻唑基、哌啶基、六氢哌啶基、吡啶基、二氢吡啶基、四氢吡啶基、噻嗪基、吡咯基、咪唑基、吡唑基、嘧啶基、哌嗪基、吗啉基、取代哌嗪基、呋喃基、吡喃基、任选地被取代基Y取代的杂环基,任选地被取代基Y取代的芳基,任选地被取代基Y取代的杂芳基等;所述取代哌嗪基是指甲基哌嗪基、乙基哌嗪基、丙基哌嗪基、二甲基哌嗪基、甲基(乙基)哌嗪基、异丙基哌嗪基、卤取代哌嗪基;卤取代哌嗪基中卤素包括氟、氯、溴、碘其中所述取代基Y为氢原子、C1-C6烷基、卤代C1-C6烷基、卤素、硝基、羟基、芳基、氨基、C1-C6烷基氨基、C3-C7环烷基氨基、二(C1-C6烷基)氨基、氰基或C3-C7环烷基等;其中所述芳香基羰基包括苯基羰基,吡啶基羰基等。进一步,优选所述的X是氧或硫。进一步,优选的,n=0-3。进一步,优选的,R1各自独立的选自:氢、卤素、羟基、氰基、硝基、氨基、C1-C5烷基、C1-C4烷氧基、C3-C6环烷基、C1-C4烷氧基羰基、C1-C5烷基羰基、氨基羰基、C1-C6烷基氨基羰基、取代氨基、恶唑基、噻唑基、哌啶基、六氢哌啶基、吡啶基、二氢吡啶基、四氢吡啶基、噻嗪基、吡咯基、咪唑基、吡唑基、嘧啶基、哌嗪基、吗啉基、取代哌嗪基、呋喃基、吡喃基。进一步,优选的,m=0-4;进一步,优选的,R2各自独立的选自:氢、氟、氯、溴、羟基、氰基、硝基、C1-C4烷基、C1-C3烷氧基、C3-C7环烷基、卤代C1-C6烷基、C2-C6烯基、羟基取代的C1-C6烷基、二(C1-C6烷基)氨基-C1-C6烷基、氨基、C1-C6烷基氨基、C3-C7环烷基氨基、二(C1-C6烷基)氨基、氨基-C1-C6烷基氨基、C1-C6烷氧基C1-C6烷基氨基、C1-C6烷氧基羰基、C1-C6烷基氨基、二(C1-C6烷氧基-C1-C6烷基)氨基、氨基羰基、C1-C6烷基氨基羰基、二(C1-C6烷基)氨基羰基、C3-C7环烷基氨基羰基、C1-C6烷氧基、C3-C7环烷氧基、羟基-C1-C6烷氧基、卤代C1-C6烷氧基、氨基C1-C6烷基、氨基C1-C6烷氧基、C1-C6烷基砜、C2-C6烯基砜、C3-C7环烷基砜、C3-C7环烷基、卤代C3-C7环烷基、哌啶基氨基、N-甲基哌啶-4-羰基、哌嗪-(C1-C6)烷基、吡咯甲酰胺基、N-甲基哌啶甲酰胺基、或杂环C1-C6烷基氧基。本发明同时还提供了一种制备上述式Ⅰ化合物的方法。合成上述氧氮杂唑酮衍生物的方法,包括以下步骤:(1)以2-羟基苯乙酮(或者苯环上具有取代基团的2-羟基苯乙酮)和取代苯甲醛为起始原料,经曼尼希缩合反应制得黄烷酮化合物1;(2)随后经扩环反应制得化合物2;(3)其中产物2a(化合物2的一种,见以下反应式)经磺化、胺化得到化合物2f;(4)另产物2d和2h(化合物2的一种,见以下反应式)为右边苯环-NO2取代,经还原Fe粉还原得到3d和/或3h;(5)3d与芳香羧酸缩合制得酰胺产物4d和/或4e。反应流程如下:Scheme1.Reagentsandconditions:a:PhNH2/I2,CH3OH,45℃,20-30h.b:NaN3,CH3COOH/H2SO4,0℃-45℃,1-2h.Scheme2.Reagentsandconditions:c:Fe/HCl,75%ethylalcohol.d:differentcarboxylicacidtreatedwiththionylchloride,DimethylFormamidecatalyst.e:DIEA(Diisopropylethylamine),CH2Cl2,ice-bath,1-2h.Scheme3.Reagentsandconditions:c1:ClSO3H,ice-bath.d1:ammoniumhydroxide,100℃本发明提供了一种具有式Ⅰ结构的全新化合物,可以有效的抑制肿瘤细胞的生长,应用于多种人体自身细胞异常生长导致的肿瘤疾病的治疗和预防。上述化合物在预防和/或治疗肿瘤疾病中的应用。与现有技术相比,本发明的有益效果:1.本发明发现了具有抗肿瘤活性的新颖结构的氧氮杂唑酮衍生物。2.为抗肿瘤新药的研发奠定基础,为广大肿瘤患者提供抗肿瘤新药。3.抗肿瘤药物具有巨大的经济效益。附图说明:图1是黄烷酮(1a)1H-NMR(400MHz,DMSO-d6)谱图。图2是黄烷酮(1a)13C-NMR(101MHz,DMSO-d6)谱图。图3是化合物(2a)1H-NMR(400MHz,DMSO-d6)谱图。图4是化合物(2a)13C-NMR(101MHz,DMSO-d6)谱图。图5是化合物(3h)1H-NMR(400MHz,DMSO-d6)谱图。图6是化合物(3h)13C-NMR(101MHz,DMSO-d6)谱图。图7是化合物(4d)1H-NMR(400MHz,DMSO-d6)谱图。图8是化合物(4d)13C-NMR(101MHz,DMSO-d6)谱图。图9是化合物(2f)13C-NMR(101MHz,DMSO-d6)谱图。具体实施方式本发明中部分专业术语解释如下:低级烷基包括甲基、乙基、丙基、异丙基、环丙基、丁基、异丁基、叔丁基、正戊基、异戊基、新戊基、己基、庚基。卤代包括氟代,氯代,溴代,碘代。C1-C5烷基包括:甲基、乙基、丙基、异丙基、丁基、异丁基、叔丁基、正-戊基、异-戊基、新戊基、仲戊基、叔戊基等。C1-C7烷基包括:甲基、乙基、丙基、异丙基、丁基、异丁基、叔丁基、正-戊基、异-戊基、新戊基、仲戊基、叔戊基、己基、庚基等。低级烯基包括:乙烯基、丙烯基、丁烯基、戊烯基、己烯基、庚烯基、辛烯基等。下面结合试验例及具体实施方式对本发明作进一步的详细描述。但不应将此理解为本发明上述主题的范围仅限于以下的实施例,凡基于本
发明内容所实现的技术均属于本发明的范围。实施例1制备式1化合物向25mL干燥的圆底烧瓶中加入20mL甲醇,依次加入苯甲醛(1.0mmol)、苯胺(1.5mmol)、2-羟基苯乙酮(1.2mmol)、I2(0.3mmol)于45℃搅拌,经曼尼希缩合反应,薄层色谱(TLC)跟踪反应进程。反应结束后,除去溶剂,加入冰的饱和Na2S2O3溶液20mL,用CH2Cl2(3*20mL)萃取。合并有机相,依次用饱和NaCl溶液、冰水洗,无水Na2SO4干燥。浓缩有机相得到粗品,环己烷-乙酸乙酯(V:V=6:1)柱色谱分离纯化。得到黄烷酮化合物(1a)。实施例2制备式2化合物向25mL干燥的圆底烧瓶中加入4.5mmol黄烷酮(1a)、6.2mmolNaN3、3.3mL冰乙酸,搅拌使溶解,冰浴冷却后,缓慢滴加0.66mL浓H2SO4。于冰浴上搅拌,直至气泡产生并继续在冰浴完成放热反应。转移至45-50℃反应45min。冷却,加入10%NaHCO3冰水溶液,调PH呈中性。乙酸乙酯萃取(3*20),无水MgSO4干燥,浓缩有机相得到粗品,环己烷-乙酸乙酯(V:V=2:1)柱色谱分离纯化。得到目标化合物(2a)。实施例3制备式2f化合物取2-苯基-3,4-二氢苯并[f][1,4]-氧氮杂卓-5-(2H)-酮(2a)2.5mmol置于25ml三口瓶中,于冰浴上加入2mlClSO3H,连接尾气处理装置,搅拌直至无气泡产生。缓慢将反应液倾入干净冰水中、不断搅拌,有固体析出,抽滤,清水洗涤滤饼。将滤饼置于反应瓶中,冰浴上缓慢加入3ml浓氨水,转移至100℃搅拌20-40min,加入15ml水,继续搅拌5-10min,静置、抽滤,滤饼干燥得目标化合物(2f)。实施例4制备式3d、3h化合物向25mL干燥的圆底烧瓶中加入25mL75%乙醇、还原8mmolFe粉,稀盐酸调节PH值3~4,80℃搅拌10min后,加入2mmol化合物2d或2h,80℃回流,薄层色谱(TLC)跟踪反应进程,反应完全后,冷却,抽滤,滤液用NaHCO3溶液调节PH值8~9,静置、抽滤,滤液用乙酸乙酯(3*20mL)萃取。合并有机相,无水MgSO4干燥,浓缩有机相得目标化合物3d、3h。实施例5制备式4d、4e化合物向25mL干燥的圆底烧瓶中加入2mmol羧酸,适量SOCl2,催化量DMF(DimethylFormamide),80℃回流,薄层色谱(TLC)跟踪反应进程,反应完全后,旋蒸除去多余SOCl2,溶加CH2Cl2溶解,缓慢滴加至冰浴冷却的含化合物3d、DIEA的CH2Cl2溶液中。薄层色谱(TLC)跟踪反应进程,反应结束后,浓缩有机相得到粗品,用环己烷-乙酸乙酯(V:V=2:1)柱色谱分离纯化。得到目标化合物4d、4e。实施例6苯并[f][1,4]氧氮杂唑酮衍生物的抗肿瘤细胞活性按照实施例1-4的工艺步骤,多次合成得到表1和表2中化合物(Coupounds)并将其用于相关的抗肿瘤细胞活性试验。将部分上述合成的化合物采用体外培养的试验方法测试其对于Hep-3B、Hela和MDA-MB-231肿瘤细胞的IC50浓度(MIC),结果如下:细胞增殖抑制实验采用MTT法:分别用完全培养液调整细胞株为Hep-3B、Hela、MDA-MB-231三种细胞细胞浓度为2×104/ml,接种于96孔板,每孔100μL,培养过夜,次日分别用不同剂量的待筛选化合物(终浓度分别为40,20,10,5,2.5,1.25μmol/L)处理细胞,同时等体积的溶剂对照组,DMSO浓度为0.1%(0.1%的DMSO对细胞增殖没有影响)。每个设5个复孔,37℃,5%CO2培养48小时后,每孔加入5mg/mlMTT试剂20μL,继续培养2~4h,弃上清液,再加DMSO150μL,振荡混匀15min,用酶标仪(λ=570nm)测定吸光度(A)值(A值与活细胞数成正比),取其平均值。相对细胞增殖抑制率(%)=(对照组A570-实验组A570)/对照组A570×100%,抑制率为50%的化合物浓度即为该化合物的IC50值。实验至少重复3次。阳性对照采用5-F脲嘧啶。根据实施例1-6的实验方法,合成的化合物,经过核磁图谱监测分析验证所得化合物,部分结果如图1-9所示。将合成得到的化合物进行肿瘤细胞活性抑制试验,部分结果见表1记载。表1部分合成化合物的活性表征结果*在表格中取代基团R1、R2前面记录的数字表述取代基团在母核结构上的连接位置,具体是在母核上对应的苯环上的位置。上述试验结果表明本发明制备的式Ⅰ结构的化合物对肿瘤细胞具有生长具有良好抑制作用,可以用于多种人体肿瘤疾病的治疗。部分化合物的谱图(1)黄烷酮中间体化合物(1a)的谱图图1是黄烷酮(1a)1H-NMR(400MHz,DMSO-d6)谱图。图2是黄烷酮(1a)13C-NMR(101MHz,DMSO-d6)谱图。(2)化合物(2a)的谱图图3是(2a)1H-NMR(400MHz,DMSO-d6)谱图。图4是化合物(2a)13C-NMR(101MHz,DMSO-d6)谱图。(3)化合物(3h)的谱图图5是化合物(3h)1H-NMR(400MHz,DMSO-d6)谱图。图6是化合物(3h)13C-NMR(101MHz,DMSO-d6)谱图。(4)化合物(4d)的谱图图7是化合物(4d)1H-NMR(400MHz,DMSO-d6)谱图。图8是化合物(4d)13C-NMR(101MHz,DMSO-d6)谱图。(5)化合物(2f)谱图图9是化合物(2f)13C-NMR(101MHz,DMSO-d6)谱图。根据本发明的实施例1-6的流程合成以下化合物成分:表2合成化合物情况CompoundR1R220012-OH3-丙基氧基20022-环己基H2003H4-吡嗪基20043-CN2-甲基、3-甲氧基20052-NH23-Br20062-乙基丙基氨基3-NHCH32007H3-(4-甲基-1-哌嗪基)20082-丙基氧基羰基H20092-哌啶基4-SO2C2H520101-甲基、2-乙基3-正戊基20113-丁基2、3位形成的5元氮杂环,咪唑环2012H4-乙氧基20132-丙基氨基4-(甲基)乙基氨基*在表格中取代基团R1、R2前面记录的数字表述取代基团的位置。当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1