新型前体药物及其使用方法与流程
技术领域
本发明涉及用作靶向细胞毒素试剂的化合物及其使用方法。在具体的实施方式中,本发明涉及硝基苯甲酰胺氮芥、硝基苯甲酰胺氮芥醇和它们相应的磷酸酯。
背景技术:
肿瘤-选择性前体药物(相对可在体内选择性转化成更有活性化合物的活性化合物而言)的使用是癌症疗法中的重要概念(见例如,Denny,Eur.J:Med.Chem.(2001)36,577)。例如前体药物可在酶的作用下转化成抗肿瘤剂,所述酶可与结合肿瘤相关的抗原的单克隆抗体结合。这种前体药物与这种酶/单克隆抗体缀合物的组合表示非常有力的临床试剂。这种通常称为“抗体导向的酶前体药物疗法”(ADEPT)的癌症疗法的方法公开在国际公开WO/1988/007378中。
称为“病毒导向酶前体药物疗法”(VDEPT)的进一步治疗方法已经被推荐为使用前体药物治疗患者中肿瘤细胞的方法。用携带编码能够活化前体药物的酶的基因的病毒载体靶向肿瘤细胞。基因可通过组织特异性启动子或增强子序列转录调节。病毒载体进入肿瘤细胞并且表达酶,以便前体药物在肿瘤细胞中转化成活性药物(Huber等,Proc.Natl.Acad.Sci.USA(1991)88,8039)。可选地,已经使用用于递送基因的非病毒方法。这种方法,其包括磷酸钙共沉淀、微注射、脂质体、直接DNA吸收和受体介导的DNA-转移。这些在Morgan&French,Annu.Rev.Bioehem.,1993,62;191中综述。术语“GDEPT”(基因导向的酶前体药物疗法)用于包括病毒和非病毒递送系统二者(Denny等US 6,310,237)。非病毒递送系统的一个例子是肿瘤寄生菌梭菌,其用于称为梭菌导向的酶前体药物疗法(CDEPT)的方法中。
可通过哺乳类黄素蛋白酶和细菌黄素蛋白酶还原许多硝基芳族化合物,所述酶有效逐步添加多达六个电子。主要酶代谢物通常是4-电子还原的物质(羟胺)。许多硝基苯基氮芥和硝基苯基氮丙啶已经报道作为结合硝基还原酶用于基因导向的酶前体药物疗法(GDEPT)中的前体药物。尤其,CB 1954[5-(吖丙啶-1-基)-2,4-二硝基苯甲酰胺](化合物1,方案1)报道为是分离自大肠杆菌的需氧菌硝基还原酶NTR(nfsB基因产物)的底物(Boland等,Biochem.Pharmacol.1991,41,867-875;Anlezark等,Biochem.Pharmacol,15,1992,44,2289-2295;Parkinson等,J.Med.Chem.2000,43,3624)。该化合物已经用作ADEPT(Knox等,Biochem。Pharmacol.,1995,49,1641-1647)和GDEPT(Bridgewater等,Eur.J.Cancer,1995,31A,2362-2370;Bailey等,Gene Ther.,1996,3,1143-1150;Bailey和Hart,Gene Ther.,1997,4,80-81;Green等,Cancer Gene Ther.,1997,4,229-238)应用二者中,包括临床试验(Chung-Faye等,Clin.Cancer Res.,2001,7,2662-2668)的前药。类似地,二硝基苯基氮芥SN23862(化合物2,方案1)也是大肠杆菌NfsB的底物,并且显示对表达酶的细胞系的选择性毒性。其被硝基基团还原活化(Palmer等,J.Med.Chem.,1995,38,1229;Kestell等,Cancer Chemother.Pharmacol.,2000,46,365-374)。4-SO2Me衍生物(化合物3,方案1)也是底物(Atwell等,Anti-Cancer Drug Des.,1996,11,553),也是二溴氮芥类似物(化合物4,方案1)(Atwell等,J.Med.Chem.,2007,50,1197-1212)。前药1-4(方案1)具有差的水性溶解度。例如,为了测定前药4在具有异种移植物裸鼠中的效力,其施用在洁净的DMSO或DMSO/聚乙二醇/水中(Atwell等,J.Med.Chem.,2007,50,1197-1212),产生最大耐受剂量的很大变化。
方案1
为了溶解化合物的目的,已经描述了氮芥的一些磷酸类似物。最佳已知的是雌莫司汀磷酸盐,其已经显示为结合各种微管相关蛋白上的微管蛋白结合结构域(Moraga等,Biochim.Biophys.Acta,1992,1121,97-103),并且其已经显示在晚期乳腺癌中是有活性的(Keren-Rosenberg等,Semin.Oncol.,1997,24(Suppl.3),26-29),但是未显示被NTR活化。
在甲酰胺(-CONH-)基团具有二硝基苯甲酰胺氮芥的醇侧链侧基和它们的磷酸衍生物描述为GDEPT应用的生物还原性药物(WO/2008/030112和WO/2005/042471)。本公开的中心是提供以基本上最小旁观者效应的细胞剔除的前体药物,所述旁观者效应是用于描述来自表达细菌硝基还原酶的靶细胞的细胞毒素代谢分子的背扩散,以剔除细菌硝基还原酶天然细胞。没有提供旁观者效率数据。
使邻近细胞消毒以其他方式不能活化靶向细胞毒素试剂的能力对于结合硝基还原酶的试剂活性具有重要的作用。在比如GDEPT、VDEPT、CDEPT和ADEPT的方法中使用的基因/酶递送技术固有地是异质的,使得必须有效再分配活化的细胞毒素代谢分子,以抑制更大量的邻近细胞。因此,旁观者效应是通过产生局部扩散以剔除邻近载体天然细胞的细胞毒素代谢分子,补偿该预期的异质性的重要机制。
除了被外源性氧非依赖性两电子硝基还原酶活化,期望设计硝基芳族前体药物,其具有适当的电子亲和性的硝基取代基,能够被内源性人单电子还原酶还原,以生产可容易被分子氧反向氧化的硝基自由基阴离子。在身体充分结合氧的组织中,亲本前体药物在无效氧化还原循环中再形成,但是在存在人实体瘤中出现的病理学低氧的情况下,能够发生净还原成羟胺和胺细胞毒素代谢分子,提供肿瘤选择性细胞杀伤。这种化合物称为低氧-活化前体药物(HAP)或低氧选择性细胞毒素(HSC)。
II期临床候选物PR-104是3,5-二硝基苯甲酰胺水溶性磷酸前体药物,其被全身性磷酸酶水解之后,释放‘低氧-活化’和‘细菌硝基还原酶-活化’前体药物PR-104A。通过肿瘤的缺氧细胞中内源人单电子还原酶或通过外源性氧非依赖性两电子硝基还原酶,比如肿瘤中表达的基因工程化的细菌硝基还原酶代谢PR-104A产生DNA交联氮芥细胞毒素代谢分子PR-104H和PR-104M(方案2)(Patterson等,Clin Can Res 2007,13:3922-32)。
2e-细菌硝基还原酶或人AKR1C3
方案2
出人意料地PR-104A通过称为醇醛-酮还原酶1C3(AKR1C3)的内源人还原酶也经受2e-还原。该需氧通路产生相同的细胞毒素代谢分子。人CD34+髓性祖细胞中AKR1C3的表达可导致缺少PR-104相对于正常骨髓对实体瘤的选择性,损害PR-104的治疗指数。所以,期望消除这种通过AKR1C3的PR-104的脱原理需氧活化。
本发明的目的是提供基本上不被人AKR1C3酶活化的一个或多个前体药物,或至少为公众提供有用选择。
技术实现要素:
在本发明的第一方面中,提供了式(I)的化合物
其中W表示Cl、Br、I、OSO2R,
x表示Cl、Br、I、OSO2R,
Y表示H、CN、SO2R,
每个R独立地表示低级C1-6烷基基团,
Z选自式(Ia)的任何自由基,或所述化合物药学上可接受的盐
其中
R1表示H,或低级C1-6烷基基团;
R2和R3可独立地表示H,或低级C1-6烷基基团,或
R2和R3可连接在一起形成取代的或未取代的包含5元或6元的杂环;
n表示2至6;
*表示与式I附接的点。
在第一方面的一个具体实施方式中,本发明提供了式(Ib)的化合物
其中Y表示H、CN、SO2R,
R表示甲基或乙基基团,
Z选自式(Ic)的任何自由基,或所述化合物药学上可接受的盐
其中
R1表示H,或低级C1-6烷基基团;
R2和R3可独立地表示H,或低级C1-6烷基基团,或
R2和R3可连接在一起形成取代的或未取代的包含5元或6元的杂环;
n表示2至6;
*表示与式Ib附接的点。
在一种实施方式中,式(I)的化合物包括式(Id)表示的化合物,
其中n表示2至6,
W表示Cl、Br、I、OSO2R,
X表示Cl、Br、I、OSO2R,
每个R独立地表示低级C1-6烷基基团,和
R1表示H,或低级C1-6烷基基团。
在另一实施方式中,W是溴或碘。
在另一实施方式中,X是溴或OSO2Me。
在另一实施方式中,R是甲基或乙基。
在另一实施方式中,R1是氢、甲基或乙基。
在另一实施方式中,n表示2或3
在具体的实施方式中,式(I)的化合物包括式(Ie)表示的化合物,
其中n表示2至6,和
R表示甲基或乙基。
在另一实施方式中,式(I)的化合物选自:
2-(5-(双(2-溴乙基)氨基)-N-甲基-4-(甲基磺酰基)-2-硝基苯甲酰氨基)乙基二氢磷酸酯(化合物10),
2-(5-(双(2-溴乙基)氨基)-4-(乙磺酰基)-N-甲基-2-硝基苯甲酰氨基)乙基二氢磷酸酯(化合物11),
3-(5-(双(2-溴乙基)氨基)-N-甲基-4-(甲基磺酰基)-2-硝基苯甲酰氨基)丙基二氢磷酸酯(化合物12),
3-(5-(双(2-溴乙基)氨基)-4-(乙磺酰基)-N-甲基-2-硝基苯甲酰氨基)丙基二氢磷酸酯(化合物13),
2-(5-(双(2-溴乙基)氨基)-4-氰基-N-甲基-2-硝基苯甲酰氨基)乙基二氢磷酸酯(化合物69),
3-(5-(双(2-溴乙基)氨基)-4-氰基-N-甲基-2-硝基苯甲酰氨基)丙基二氢磷酸酯(化合物70),
2-(5-(双(2-溴乙基)氨基)-4-(甲基磺酰基)-2-硝基苯甲酰氨基)乙基二氢磷酸酯(化合物300),
3-(5-(双(2-溴乙基)氨基)-4-(甲基磺酰基)-2-硝基苯甲酰氨基)丙基二氢磷酸酯(化合物308),
3-(5-(双(2-溴乙基)氨基)-4-(乙磺酰基)-2-硝基苯甲酰氨基)丙基二氢磷酸酯(化合物309),
2-((2-溴乙基)(2-氰基-5-(甲基(2-(膦酰氧基)乙基)氨甲酰基)-4-硝基苯基)氨基)乙基甲磺酸酯(化合物350),和
2-((2-溴乙基)(2-氰基-5-(甲基(3-(膦酰氧基)丙基)氨甲酰基)-4-硝基苯基)氨基)乙基甲磺酸酯(化合物351)。
在另一实施方式中,式(I)的化合物选自式(If)表示的化合物,
其中n表示2至6,
W表示Cl、Br、I、OSO2R,
X表示Cl、Br、I、OSO2R,
每个R独立地表示低级C1-6烷基基团,和
R1表示H,或低级C1-6烷基基团。
在另一实施方式中,W是溴或碘。
在另一实施方式中,X是溴或OSO2Me。
在另一实施方式中,R是甲基或乙基。
在另一实施方式中,R1是氢、甲基或乙基。
在另一实施方式中,n表示2或3
在具体的实施方式中,式(I)的化合物选自式(Ig)表示的化合物,
其中n表示2至6,
R表示甲基或乙基。
在另一实施方式中,式(I)的化合物选自:
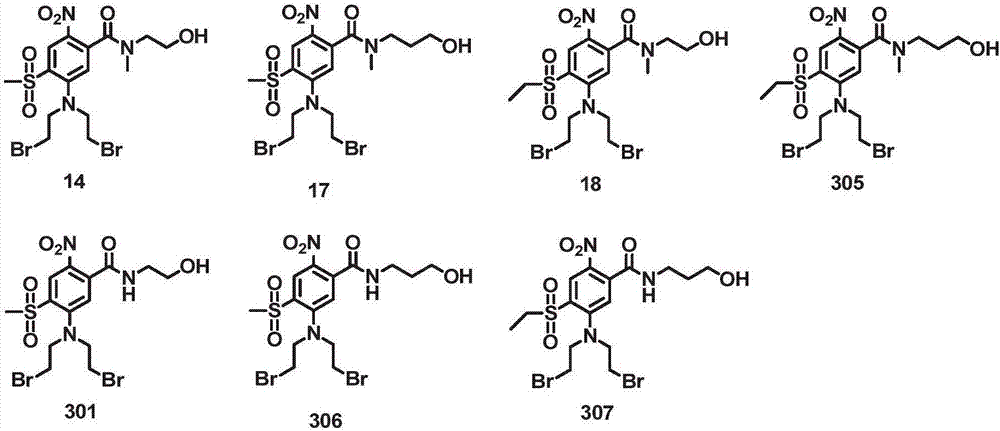
5-(双(2-溴乙基)氨基)-N-(2-羟乙基)-N-甲基-4-(甲基磺酰基)-2-硝基苯甲酰胺(化合物14),
5-(双(2-溴乙基)氨基)-N-(3-羟丙基)-N-甲基-4-(甲基磺酰基)-2-硝基苯甲酰胺(化合物17),
5-(双(2-溴乙基)氨基)-4-(乙磺酰基)-N-(2-羟乙基)-N-甲基-2-硝基苯甲酰胺(化合物18),
5-(双(2-溴乙基)氨基)-4-氰基-N-(2-羟乙基)-N-甲基-2-硝基苯甲酰胺(化合物67),
5-(双(2-溴乙基)氨基)-4-氰基-N-(3-羟丙基)-N-甲基-2-硝基苯甲酰胺(化合物68),
5-(双(2-溴乙基)氨基)-N-(2-羟乙基)-4-(甲基磺酰基)-2-硝基苯甲酰胺(化合物301),
5-(双(2-溴乙基)氨基)-4-(乙磺酰基)-N-(3-羟丙基)-N-甲基-2-硝基苯甲酰胺(化合物305),
5-(双(2-溴乙基)氨基)-N-(3-羟丙基)-4-(甲基磺酰基)-2-硝基苯甲酰胺(化合物306),
5-(双(2-溴乙基)氨基)-4-(乙磺酰基)-N-(3-羟丙基)-2-硝基苯甲酰胺(化合物307),
2-((2-溴乙基)(2-氰基-5-((2-羟乙基)(甲基)氨甲酰基)-4-硝基苯基)氨基)乙基甲磺酸酯(化合物348),和
2-((2-溴乙基)(2-氰基-5-((3-羟丙基)(甲基)氨甲酰基)-4-硝基苯基)氨基)乙基甲磺酸酯(化合物349)。
在另一实施方式中,式(I)的化合物选自式(Ih)表示的化合物,
其中W表示Cl、Br、I、OSO2R,
X表示Cl、Br、I、OSO2R,
每个R独立地表示低级C1-6烷基基团,
R1表示H,或低级C1-6烷基基团。
在另一实施方式中,W是溴或碘。
在另一实施方式中,X是溴或OSO2Me。
在另一实施方式中,R是甲基或乙基。
在另一实施方式中,R1是甲基、乙基,丙基或异丙基。
在另一实施方式中,式(I)的化合物选自式(Ii)表示的化合物,
其中R表示低级C1-6烷基基团,
R1表示H,或低级C1-6烷基基团。
在另一实施方式中,R是甲基或乙基。
在另一实施方式中,R1是甲基或乙基。
在另一实施方式中,上面式(I)定义的化合物选自:
(5-(双(2-溴乙基)氨基)-4-(甲基磺酰基)-2-硝基苯基)(4-甲基哌嗪-1-基)甲酮(化合物22),
(5-(双(2-溴乙基)氨基)-4-(甲基磺酰基)-2-硝基苯基)(4-乙基哌嗪-1-基)甲酮(化合物23),
(5-(双(2-溴乙基)氨基)-4-(甲基磺酰基)-2-硝基苯基)(4-异丙基哌嗪-1-基)甲酮(化合物24),
(5-(双(2-溴乙基)氨基)-4-(乙磺酰基)-2-硝基苯基)(4-甲基哌嗪-1-基)甲酮(化合物25),
(5-(双(2-溴乙基)氨基)-4-(乙磺酰基)-2-硝基苯基)(4-乙基哌嗪-1-基)甲酮(化合物26),
(5-(双(2-溴乙基)氨基)-4-(乙磺酰基)-2-硝基苯基)(4-异丙基哌嗪-1-基)甲酮(化合物27),
5-(双(2-溴乙基)氨基)-4-(甲基磺酰基)-N-(2-吗啉乙基)-2-硝基苯甲酰胺(化合物28),
5-(双(2-溴乙基)氨基)-N-甲基-4-(甲基磺酰基)-N-(2-吗啉乙基)-2-硝基苯甲酰胺(化合物29),
5-(双(2-溴乙基)氨基)-4-(甲基磺酰基)-N-(3-吗啉丙基)-2-硝基苯甲酰胺(化合物30),
5-(双(2-溴乙基)氨基)-N-甲基-4-(甲基磺酰基)-N-(3-吗啉丙基)-2-硝基苯甲酰胺(化合物31),
5-(双(2-溴乙基)氨基)-N-(2-(4-甲基哌嗪-1-基)乙基)-4-(甲基磺酰基)-2-硝基苯甲酰胺(化合物32),
5-(双(2-溴乙基)氨基)-N-甲基-N-(2-(4-甲基哌嗪-1-基)乙基)-4-(甲基磺酰基)-2-硝基苯甲酰胺(化合物33),
5-(双(2-溴乙基)氨基)-N-(3-(4-甲基哌嗪-1-基)丙基)-4-(甲基磺酰基)-2-硝基苯甲酰胺(化合物34),
5-(双(2-溴乙基)氨基)-N-甲基-N-(3-(4-甲基哌嗪-1-基)丙基)-4-(甲基磺酰基)-2-硝基苯甲酰胺(化合物35),
5-(双(2-溴乙基)氨基)-N-(2-(二甲氨基)乙基)-4-(甲基磺酰基)-2-硝基苯甲酰胺(化合物36),
5-(双(2-溴乙基)氨基)-N-(2-(二甲氨基)乙基)-N-甲基-4-(甲基磺酰基)-2-硝基苯甲酰胺(化合物37),
5-(双(2-溴乙基)氨基)-N-(3-(二甲氨基)丙基)-4-(甲基磺酰基)-2-硝基苯甲酰胺(化合物38),
5-(双(2-溴乙基)氨基)-N-(3-(二甲氨基)丙基)-N-甲基-4-(甲基磺酰基)-2-硝基苯甲酰胺(化合物39),
5-(双(2-溴乙基)氨基)-4-(乙磺酰基)-N-(2-吗啉乙基)-2-硝基苯甲酰胺(化合物40),
5-(双(2-溴乙基)氨基)-4-(乙磺酰基)-N-甲基-N-(2-吗啉乙基)-2-硝基苯甲酰胺(化合物41),
5-(双(2-溴乙基)氨基)-4-(乙磺酰基)-N-(3-吗啉丙基)-2-硝基苯甲酰胺(化合物42),
5-(双(2-溴乙基)氨基)-4-(乙磺酰基)-N-甲基-N-(3-吗啉丙基)-2-硝基苯甲酰胺(化合物43),
5-(双(2-溴乙基)氨基)-4-(乙磺酰基)-N-(2-(4-甲基哌嗪-1-基)乙基)-2-硝基苯甲酰胺(化合物44),
5-(双(2-溴乙基)氨基)-4-(乙磺酰基)-N-甲基-N-(2-(4-甲基哌嗪-1-基)乙基)-2-硝基苯甲酰胺(化合物45),
5-(双(2-溴乙基)氨基)-4-(乙磺酰基)-N-(3-(4-甲基哌嗪-1-基)丙基)-2-硝基苯甲酰胺(化合物46),
5-(双(2-溴乙基)氨基)-4-(乙磺酰基)-N-甲基-N-(3-(4-甲基哌嗪-1-基)丙基)-2-硝基苯甲酰胺(化合物47),
5-(双(2-溴乙基)氨基)-N-(2-(二甲氨基)乙基)-4-(乙磺酰基)-2-硝基苯甲酰胺(化合物48),
5-(双(2-溴乙基)氨基)-N-(2-(二甲氨基)乙基)-4-(乙磺酰基)-N-甲基-2-硝基苯甲酰胺(化合物49),
5-(双(2-溴乙基)氨基)-N-(3-(二甲氨基)丙基)-4-(乙磺酰基)-2-硝基苯甲酰胺(化合物50),
5-(双(2-溴乙基)氨基)-N-(3-(二甲氨基)丙基)-4-(乙磺酰基)-N-甲基-2-硝基苯甲酰胺(化合物51),
3-(5-(双(2-溴乙基)氨基)-4-(甲基磺酰基)-2-硝基苯甲酰氨基)丙酸(化合物52),
4-(5-(双(2-溴乙基)氨基)-4-(甲基磺酰基)-2-硝基苯甲酰氨基)丁酸(化合物53),
3-(5-(双(2-溴乙基)氨基)-N-甲基-4-(甲基磺酰基)-2-硝基苯甲酰氨基)丙酸(化合物54),
4-(5-(双(2-溴乙基)氨基)-N-甲基-4-(甲基磺酰基)-2-硝基苯甲酰氨基)丁酸(化合物55),
3-(5-(双(2-溴乙基)氨基)-4-(乙磺酰基)-2-硝基苯甲酰氨基)丙酸(化合物56),
4-(5-(双(2-溴乙基)氨基)-4-(乙磺酰基)-2-硝基苯甲酰氨基)丁酸(化合物57),
3-(5-(双(2-溴乙基)氨基)-4-(乙磺酰基)-N-甲基-2-硝基苯甲酰氨基)丙酸(化合物58),
4-(5-(双(2-溴乙基)氨基)-4-(乙磺酰基)-N-甲基-2-硝基苯甲酰氨基)丁酸(化合物59)
2-(双(2-溴乙基)氨基)-4-(4-甲基哌嗪-1-羰基)-5-硝基苯甲腈(化合物71),
2-(双(2-溴乙基)氨基)-4-(4-乙基哌嗪-1-羰基)-5-硝基苯甲腈(化合物72),
2-(双(2-溴乙基)氨基)-4-(4-异丙基哌嗪-1-羰基)-5-硝基苯甲腈(化合物73),
3-(5-(双(2-溴乙基)氨基)-4-氰基-2-硝基苯甲酰氨基)丙酸(化合物74),
4-(5-(双(2-溴乙基)氨基)-4-氰基-2-硝基苯甲酰氨基)丁酸(化合物75),
3-(5-(双(2-溴乙基)氨基)-4-氰基-N-甲基-2-硝基苯甲酰氨基)丙酸(化合物76),
4-(5-(双(2-溴乙基)氨基)-4-氰基-N-甲基-2-硝基苯甲酰氨基)丁酸(化合物77),
5-(双(2-溴乙基)氨基)-4-氰基-N-(2-吗啉乙基)-2-硝基苯甲酰胺(化合物78),
5-(双(2-溴乙基)氨基)-4-氰基-N-甲基-N-(2-吗啉乙基)-2-硝基苯甲酰胺(化合物79),
5-(双(2-溴乙基)氨基)-4-氰基-N-(3-吗啉丙基)-2-硝基苯甲酰胺(化合物80),
5-(双(2-溴乙基)氨基)-4-氰基-N-甲基-N-(3-吗啉丙基)-2-硝基苯甲酰胺(化合物81),
5-(双(2-溴乙基)氨基)-4-氰基-N-(2-(4-甲基哌嗪-1-基)乙基)-2-硝基苯甲酰胺(化合物82),
5-(双(2-溴乙基)氨基)-4-氰基-N-甲基-N-(2-(4-甲基哌嗪-1-基)乙基)-2-硝基苯甲酰胺(化合物83),
5-(双(2-溴乙基)氨基)-4-氰基-N-(3-(4-甲基哌嗪-1-基)丙基)-2-硝基苯甲酰胺(化合物84),
5-(双(2-溴乙基)氨基)-4-氰基-N-甲基-N-(3-(4-甲基哌嗪-1-基)丙基)-2-硝基苯甲酰胺(化合物85),
5-(双(2-溴乙基)氨基)-4-氰基-N-(2-(二甲氨基)乙基)-2-硝基苯甲酰胺(化合物86),
5-(双(2-溴乙基)氨基)-4-氰基-N-(2-(二甲氨基)乙基)-N-甲基-2-硝基苯甲酰胺(化合物87),
5-(双(2-溴乙基)氨基)-4-氰基-N-(3-(二甲氨基)丙基)-2-硝基苯甲酰胺(化合物88),
5-(双(2-溴乙基)氨基)-4-氰基-N-(3-(二甲氨基)丙基)-N-甲基-2-硝基苯甲酰胺(化合物89),
2-((2-溴乙基)(5-(4-甲基哌嗪-1-羰基)-2-(甲基磺酰基)-4-硝基苯基)氨基)乙基甲磺酸酯(化合物310),
2-((2-溴乙基)(5-(4-乙基哌嗪-1-羰基)-2-(甲基磺酰基)-4-硝基苯基)氨基)乙基甲磺酸酯(化合物311),
2-((2-溴乙基)(5-(4-异丙基哌嗪-1-羰基)-2-(甲基磺酰基)-4-硝基苯基)氨基)乙基甲磺酸酯(化合物312),
2-((2-溴乙基)(2-(乙磺酰基)-5-(4-甲基哌嗪-1-羰基)-4-硝基苯基)氨基)乙基甲磺酸酯(化合物313),
2-((2-溴乙基)(5-(4-乙基哌嗪-1-羰基)-2-(乙磺酰基)-4-硝基苯基)氨基)乙基甲磺酸酯(化合物314),
2-((2-溴乙基)(2-(乙磺酰基)-5-(4-异丙基哌嗪-1-羰基)-4-硝基苯基)氨基)乙基甲磺酸酯(化合物315),
2-((2-溴乙基)(2-(甲基磺酰基)-5-((2-吗啉乙基)氨甲酰基)-4-硝基苯基)氨基)乙基甲磺酸酯(化合物316),
2-((2-溴乙基)(5-(甲基(2-吗啉乙基)氨甲酰基)-2-(甲基磺酰基)-4-硝基苯基)氨基)乙基甲磺酸酯(化合物317),
2-((2-溴乙基)(2-(甲基磺酰基)-5-((3-吗啉丙基)氨甲酰基)-4-硝基苯基)氨基)乙基甲磺酸酯(化合物318),
2-((2-溴乙基)(5-(甲基(3-吗啉丙基)氨甲酰基)-2-(甲基磺酰基)-4-硝基苯基)氨基)乙基甲磺酸酯(化合物319),
2-((2-溴乙基)(5-((2-(4-甲基哌嗪-1-基)乙基)氨甲酰基)-2-(甲基磺酰基)-4-硝基苯基)氨基)乙基甲磺酸酯(化合物320),
2-((2-溴乙基)(5-(甲基(2-(4-甲基哌嗪-1-基)乙基)氨甲酰基)-2-(甲基磺酰基)-4-硝基苯基)氨基)乙基甲磺酸酯(化合物321),
2-((2-溴乙基)(5-((3-(4-甲基哌嗪-1-基)丙基)氨甲酰基)-2-(甲基磺酰基)-4-硝基苯基)氨基)乙基甲磺酸酯(化合物322),
2-((2-溴乙基)(5-(甲基(3-(4-甲基哌嗪-1-基)丙基)氨甲酰基)-2-(甲基磺酰基)-4-硝基苯基)氨基)乙基甲磺酸酯(化合物323),
2-((2-溴乙基)(5-((2-(二甲氨基)乙基)氨甲酰基)-2-(甲基磺酰基)-4-硝基苯基)氨基)乙基甲磺酸酯(化合物324),
2-((2-溴乙基)(5-((2-(二甲氨基)乙基)(甲基)氨甲酰基)-2-(甲基磺酰基)-4-硝基苯基)氨基)乙基甲磺酸酯(化合物325),
2-((2-溴乙基)(5-((3-(二甲氨基)丙基)氨甲酰基)-2-(甲基磺酰基)-4-硝基苯基)氨基)乙基甲磺酸酯(化合物326),
2-((2-溴乙基)(5-((3-(二甲氨基)丙基)(甲基)氨甲酰基)-2-(甲基磺酰基)-4-硝基苯基)氨基)乙基甲磺酸酯(化合物327),
2-((2-溴乙基)(2-(乙磺酰基)-5-((2-吗啉乙基)氨甲酰基)-4-硝基苯基)氨基)乙基甲磺酸酯(化合物328),
2-((2-溴乙基)(2-(乙磺酰基)-5-(甲基(2-吗啉乙基)氨甲酰基)-4-硝基苯基)氨基)乙基甲磺酸酯(化合物329),
2-((2-溴乙基)(2-(乙磺酰基)-5-((3-吗啉丙基)氨甲酰基)-4-硝基苯基)氨基)乙基甲磺酸酯(化合物330),
2-((2-溴乙基)(2-(乙磺酰基)-5-(甲基(3-吗啉丙基)氨甲酰基)-4-硝基苯基)氨基)乙基甲磺酸酯(化合物331),
2-((2-溴乙基)(2-(乙磺酰基)-5-((2-(4-甲基哌嗪-1-基)乙基)氨甲酰基)-4-硝基苯基)氨基)乙基甲磺酸酯(化合物332),
2-((2-溴乙基)(2-(乙磺酰基)-5-(甲基(2-(4-甲基哌嗪-1-基)乙基)氨甲酰基)-4-硝基苯基)氨基)乙基甲磺酸酯(化合物333),
2-((2-溴乙基)(2-(乙磺酰基)-5-((3-(4-甲基哌嗪-1-基)丙基)氨甲酰基)-4-硝基苯基)氨基)乙基甲磺酸酯(化合物334),
2-((2-溴乙基)(2-(乙磺酰基)-5-(甲基(3-(4-甲基哌嗪-1-基)丙基)氨甲酰基)-4-硝基苯基)氨基)乙基甲磺酸酯(化合物335),
2-((2-溴乙基)(5-((2-(二甲氨基)乙基)氨甲酰基)-2-(乙磺酰基)-4-硝基苯基)氨基)乙基甲磺酸酯(化合物336),
2-((2-溴乙基)(5-((2-(二甲氨基)乙基)(甲基)氨甲酰基)-2-(乙磺酰基)-4-硝基苯基)氨基)乙基甲磺酸酯(化合物337),
2-((2-溴乙基)(5-((3-(二甲氨基)丙基)氨甲酰基)-2-(乙磺酰基)-4-硝基苯基)氨基)乙基甲磺酸酯(化合物338),
2-((2-溴乙基)(5-((3-(二甲氨基)丙基)(甲基)氨甲酰基)-2-(乙磺酰基)-4-硝基苯基)氨基)乙基甲磺酸酯(化合物339),
3-(5-((2-溴乙基)(2-((甲基磺酰基)氧)乙基)氨基)-4-(甲基磺酰基)-2-硝基苯甲酰氨基)丙酸(化合物340),
4-(5-((2-溴乙基)(2-((甲基磺酰基)氧)乙基)氨基)-4-(甲基磺酰基)-2-硝基苯甲酰氨基)丁酸(化合物341),
3-(5-((2-溴乙基)(2-((甲基磺酰基)氧)乙基)氨基)-N-甲基-4-(甲基磺酰基)-2-硝基苯甲酰氨基)丙酸(化合物342),
4-(5-((2-溴乙基)(2-((甲基磺酰基)氧)乙基)氨基)-N-甲基-4-(甲基磺酰基)-2-硝基苯甲酰氨基)丁酸(化合物343),
3-(5-((2-溴乙基)(2-((甲基磺酰基)氧)乙基)氨基)-4-(乙磺酰基)-2-硝基苯甲酰氨基)丙酸(化合物344),
4-(5-((2-溴乙基)(2-((甲基磺酰基)氧)乙基)氨基)-4-(乙磺酰基)-2-硝基苯甲酰氨基)丁酸(化合物345),
3-(5-((2-溴乙基)(2-((甲基磺酰基)氧)乙基)氨基)-4-(乙磺酰基)-N-甲基-2-硝基苯甲酰氨基)丙酸(化合物346),
4-(5-((2-溴乙基)(2-((甲基磺酰基)氧)乙基)氨基)-4-(乙磺酰基)-N-甲基-2-硝基苯甲酰氨基)丁酸(化合物347),
2-((2-溴乙基)(2-氰基-5-(4-甲基哌嗪-1-羰基)-4-硝基苯基)氨基)乙基甲磺酸酯(化合物352),
2-((2-溴乙基)(2-氰基-5-(4-乙基哌嗪-1-羰基)-4-硝基苯基)氨基)乙基甲磺酸酯(化合物353),
2-((2-溴乙基)(2-氰基-5-(4-异丙基哌嗪-1-羰基)-4-硝基苯基)氨基)乙基甲磺酸酯(化合物354),
3-(5-((2-溴乙基)(2-((甲基磺酰基)氧)乙基)氨基)-4-氰基-2-硝基苯甲酰氨基)丙酸(化合物355),
4-(5-((2-溴乙基)(2-((甲基磺酰基)氧)乙基)氨基)-4-氰基-2-硝基苯甲酰氨基)丁酸(化合物356),
3-(5-((2-溴乙基)(2-((甲基磺酰基)氧)乙基)氨基)-4-氰基-N-甲基-2-硝基苯甲酰氨基)丙酸(化合物357),
4-(5-((2-溴乙基)(2-((甲基磺酰基)氧)乙基)氨基)-4-氰基-N-甲基-2-硝基苯甲酰氨基)丁酸(化合物358),
2-((2-溴乙基)(2-氰基-5-((2-吗啉乙基)氨甲酰基)-4-硝基苯基)氨基)乙基甲磺酸酯(化合物359),
2-((2-溴乙基)(2-氰基-5-(甲基(2-吗啉乙基)氨甲酰基)-4-硝基苯基)氨基)乙基甲磺酸酯(化合物360),
2-((2-溴乙基)(2-氰基-5-((3-吗啉丙基)氨甲酰基)-4-硝基苯基)氨基)乙基甲磺酸酯(化合物361),
2-((2-溴乙基)(2-氰基-5-(甲基(3-吗啉丙基)氨甲酰基)-4-硝基苯基)氨基)乙基甲磺酸酯(化合物362),
2-((2-溴乙基)(2-氰基-5-((2-(4-甲基哌嗪-1-基)乙基)氨甲酰基)-4-硝基苯基)氨基)乙基甲磺酸酯(化合物363),
2-((2-溴乙基)(2-氰基-5-(甲基(2-(4-甲基哌嗪-1-基)乙基)氨甲酰基)-4-硝基苯基)氨基)乙基甲磺酸酯(化合物364),
2-((2-溴乙基)(2-氰基-5-((3-(4-甲基哌嗪-1-基)丙基)氨甲酰基)-4-硝基苯基)氨基)乙基甲磺酸酯(化合物365),
2-((2-溴乙基)(2-氰基-5-(甲基(3-(4-甲基哌嗪-1-基)丙基)氨甲酰基)-4-硝基苯基)氨基)乙基甲磺酸酯(化合物366),
2-((2-溴乙基)(2-氰基-5-((2-(二甲氨基)乙基)氨甲酰基)-4-硝基苯基)氨基)乙基甲磺酸酯(化合物367),
2-((2-溴乙基)(2-氰基-5-((2-(二甲氨基)乙基)(甲基)氨甲酰基)-4-硝基苯基)氨基)乙基甲磺酸酯(化合物368),
2-((2-溴乙基)(2-氰基-5-((3-(二甲氨基)丙基)氨甲酰基)-4-硝基苯基)氨基)乙基甲磺酸酯(化合物369),和
2-((2-溴乙基)(2-氰基-5-((3-(二甲氨基)丙基)(甲基)氨甲酰基)-4-硝基苯基)氨基)乙基甲磺酸酯(化合物370)。
在本发明的第二方面中,提供了式(II)的化合物
其中W表示Cl、Br、I、OSO2R,
X表示Cl、Br、I、OSO2R,
每个R独立地表示低级C1-6烷基基团,
Z选自式(IIa)的任何自由基,或其药学上可接受的盐
其中
R1表示H,或低级C1-6烷基基团,
n表示2至6
*表示与式II附接的点。
在第二方面的具体实施方式中,本发明提供了式(IIb)的化合物,或其药学上可接受的盐
其中R表示低级C1-6烷基基团,
Z选自式(IIc)的任何自由基
其中
n表示2至6
*表示与式IIb附接的点。
在一种实施方式中,式(II)的化合物包括由式(IId)表示的化合物,
其中n表示2至6,
W表示Cl、Br、I、OSO2R,
X表示Cl、Br、I、OSO2R,
R表示低级C1-6烷基基团,
R1表示H,或低级C1-6烷基基团。
在另一实施方式中,W是溴或碘。
在另一实施方式中,X是溴或OSO2Me。
在另一实施方式中,R1是氢。
在另一实施方式中,n表示2或3
在一种实施方式中,式(II)的化合物选自式(IIe)表示的化合物,
其中n表示2至6。
在另一实施方式中,式(II)的化合物选自:
2-((2-溴乙基)(4-硝基-2-((2-(膦酰氧基)乙基)氨甲酰基)苯基)氨基)乙基甲磺酸酯(化合物60),
2-((2-溴乙基)(4-硝基-2-((3-(膦酰氧基)丙基)氨甲酰基)苯基)氨基)乙基甲磺酸酯(化合物61),
在另一实施方式中,式(II)的化合物选自式(IIf)表示的化合物,
其中n表示2至6,
W表示Cl、Br、I、OSO2R,
X表示Cl、Br、I、OSO2R,
每个R独立地表示低级C1-6烷基基团,
R1表示H,或低级C1-6烷基基团。
在另一实施方式中,W是溴或碘。
在另一实施方式中,X是溴或OSO2Me。
在另一实施方式中,R1是氢。
在另一实施方式中,n表示2或3
在另一实施方式中,式(II)的化合物选自式(IIg)表示的化合物,
其中n表示2至6,
在另一实施方式中,n表示2或3
在另一实施方式中,所要求的式(II)的化合物选自:
2-((2-溴乙基)(2-((2-羟乙基)氨甲酰基)-4-硝基苯基)氨基)乙基甲磺酸酯(化合物64),
2-((2-溴乙基)(2-((3-羟丙基)氨甲酰基)-4-硝基苯基)氨基)乙基甲磺酸酯(化合物66),
在具体的实施方式中,当在包含2当量的碳酸氢钠的磷酸盐缓冲液(PBS)中测定时,第一或第二方面的化合物的溶解度大于约95mM。
在具体的实施方式中,当在pH=4的乳酸盐缓冲液中测定时,第一或第二方面的化合物的溶解度大于约10mM。
在第三方面中,提供了细胞剔除的方法,其包括使用第一或第二方面的化合物或其混合物。在具体的实施方式中,化合物是能够通过接触a)至少一种硝基还原酶,和/或b)低氧(缺氧)环境来活化的前药。
在具体的实施方式中,细胞剔除的方法,其包括:
a.选择基本上抗AKR1C3酶代谢的式(I)的化合物;
b.使步骤a.的化合物与以下接触,以产生能够剔除细胞的细胞毒素代谢物:
i.至少一种硝基还原酶,和/或
ii.缺氧环境;
c.使细胞接触细胞毒素代谢物;
其中式(I)的化合物是如第一方面中所定义。
在具体的实施方式中,细胞剔除的方法,其包括:
a.选择基本上抗AKR1C3酶代谢的式(II)的化合物;
b.使步骤a.的化合物与以下接触,以产生能够剔除细胞的细胞毒素代谢物:
i.至少一种硝基还原酶,和/或
ii.缺氧环境;
c.使细胞接触细胞毒素代谢物;
其中式(II)的化合物是如第二方面中所定义。
优选地,缺氧环境是肿瘤的缺氧区域。
优选地前药能够提供导致细胞剔除的显著旁观者效应。
在第三方面的一种实施方式中,细胞是受试者组织中的肿瘤细胞。
在第三方面的一种实施方式中,细胞是哺乳类的细胞。
在第三方面的一种实施方式中,细胞剔除的方法是癌症治疗的方法,其包括向受试者施用化合物。优选地,施用的化合物的量是治疗有效量的。优选地,该量在所述受试者的最大耐受剂量的约20%至100%之间。
在第三方面的具体的实施方式中,化合物结合能够代谢化合物的至少一种硝基还原酶施用至受试者。在具体的实施方式中,化合物结合疗法施用至受试者,所述疗法使得在肿瘤中,或治疗上接近肿瘤表达外源性硝基还原酶。在进一步的实施方式中,至少一种硝基还原酶由大肠杆菌的nfsB和/或nfsA基因编码或由其他细菌物种的直向同源基因编码。在具体的实施方式中,硝基还原酶是WO/2012/008860中描述的硝基还原酶,其包括突变体硝基还原酶和其中描述的硝基还原酶的功能上等同的变体。
在第三方面的具体的实施方式中,方法包括照射细胞的步骤。优选地,照射在施用前体药物之前、同时或之后进行。优选地,吸收照射的量是15格雷(Gy)。
在第三方面的具体的实施方式中,方法包括结合下述施用第一或第二方面的化合物或其混合物:GDEPT(基因导向的酶前体药物疗法)、VDEPT(病毒导向酶前体药物疗法)、CDEPT(梭菌导向的酶前体药物疗法)或ADEPT(抗体导向的酶前体药物疗法)。
在具体的实施方式中,CDEPT包括使用梭菌(Clostridia)微生物,该微生物选择性克隆肿瘤中出现的坏死。优选地,梭菌微生物是包含表达梭菌微生物的外源性硝基还原酶的一个或多个基因的重组微生物。优选地,硝基还原酶由大肠杆菌的nfsB和/或nfsA基因编码或由其他细菌物种的直向同源基因编码。
在第三方面的具体的实施方式中,方法进一步包括将化合物溶解在水溶液中。
在第四方面中,本发明提供了第一或第二方面的化合物或其混合物在制造剔除细胞的组合物中的用途。在具体的实施方式中,化合物是能够通过接触a)至少一种硝基还原酶,和/或b)低氧(缺氧)环境来活化的前药。
在第五方面中,本发明提供了如第一或第二方面中定义的化合物在制造用于治疗癌症或过度增生病况的组合物中的用途。
在第六方面中,本发明提供了如第一或第二方面中定义的化合物用于治疗癌症或过度增生病况中的用途。
在本发明的第七方面中,提供了治疗癌症或过度增生病况的方法,其中第一或第二方面的化合物或其混合物以治疗有效量施用至受试者的肿瘤细胞,或治疗上接近肿瘤细胞。
在第七方面具体的实施方式中,施用的治疗有效量是所述受试者的最大耐受剂量的约20%至100%之间。
在第七方面具体的实施方式中,第一或第二方面的化合物或其混合物结合至少一种硝基还原酶施用。在具体的实施方式中,化合物结合疗法施用至受试者,所述疗法使得在肿瘤中,或治疗上接近肿瘤表达外源性硝基还原酶。
在第七方面具体的实施方式中,至少一种硝基还原酶由大肠杆菌的nfsA或nfsB基因编码或由其他细菌物种的直向同源基因编码。在具体的实施方式中,硝基还原酶是WO/2012/008860中描述的硝基还原酶,其包括突变体硝基还原酶和其中描述的硝基还原酶的功能上等同的变体。
在具体的实施方式中,方法进一步包括通过接触硝基还原酶活化第一或第二方面的化合物。
在第七方面具体的实施方式中,第一或第二方面的化合物或其混合物结合下述施用:GDEPT(基因导向的酶前体药物疗法)、VDEPT(病毒导向酶前体药物疗法)、CDEPT(梭菌导向的酶前体药物疗法)或ADEPT(抗体导向的酶前体药物疗法)。
在具体的实施方式中,CDEPT包括使用选择性克隆肿瘤中出现的坏死的梭菌微生物。优选地,梭菌微生物是包含表达梭菌微生物的外源性硝基还原酶的一个或多个基因的重组微生物。
在第七方面具体的实施方式中,癌症治疗的方法进一步包括照射肿瘤细胞的步骤。优选地,照射在施用化合物之前、同时或之后进行。优选地,吸收照射的量是15格雷(Gy)。
在第七方面具体的实施方式中,方法进一步包括将化合物溶解在水溶液中。
在第八方面中,提供了药学组合物,其包括治疗有效量的第一或第二方面的化合物,或其混合物,和药学上可接受的赋形剂、佐剂、载体、缓冲液或稳定剂。
在具体的实施方式中,组合物在水溶液中是可溶的。优选地,当在包含2当量的碳酸氢钠的磷酸盐缓冲液(PBS)中测定时,如在组合物中出现的第一或第二方面的化合物的溶解度大于95mM。
在第九方面中,本发明提供了确定前体药物对被AKR1C3酶代谢的灵敏度的方法。发明人已经显示了第九方面的方法提供了高细胞密度MCL筛选,其令人吃惊地有效的从二维体外IC50筛选检测‘假阴性’。方法也尤其用于识别真实的AKR1C3-阴性前体药物,比如本发明的那些。
当本领域技术人员在阅读下述说明时,应以所有其新方面考虑的本发明的进一步方面将变得显而易见,下述说明提供本发明实际应用的至少一个例子。
附图说明
现参考附图仅仅作为例子描述本发明的实施方式,其中:
图1显示式I的二溴氮芥的代表性磷酸酯
图2显示式I的代表性二溴氮芥醇
图3显示式I的具有取代的哌嗪甲酰胺侧链的代表性二溴氮芥
图3.1显示式I的具有取代的哌嗪甲酰胺侧链的代表性溴甲磺酸酯氮芥
图4显示式I的具有胺侧链的代表性4-甲基磺酰基二溴氮芥
图5显示式I的具有胺侧链的代表性4-乙磺酰基二溴氮芥
图5.1显示式I的具有胺侧链的代表性4-甲基磺酰基溴甲磺酸酯氮芥
图5.2显示式I的具有胺侧链的代表性4-乙磺酰基溴甲磺酸酯氮芥
图6显示式I的具有酸侧链的代表性二溴氮芥
图6.1显示式I的具有酸侧链的代表性溴甲磺酸酯氮芥
图7显示式II的溴甲磺酸酯氮芥的代表性磷酸酯
图7.1显示式II的代表性溴甲磺酸酯氮芥醇
图7.2显示式I化合物具有胺侧链的代表性4-氰基二溴氮芥醇、磷酸酯
图7.2.1显示式I化合物具有胺侧链的代表性4-氰基溴甲磺酸酯氮芥醇、磷酸酯
图7.3显示式I的具有酸和胺侧链的代表性4-氰基二溴氮芥
图7.4显示式I的具有酸和胺侧链的代表性4-氰基溴甲磺酸酯氮芥
图8中的化合物X、XI和XII,图9.1中的化合物XII,和图9.1.1中的化合物XI和XXIII每个包括三个R基团。使用这些合成方法,连接至-OSO2取代基的两个R基团是相同的。另一R基团连接至砜-SO2基团可独立地于其他R基团而改变,但是仍在其中R表示低级C1-6烷基基团的式(I)中R限定的范围内。
图8显示合成式I的4-烷基砜前体药物的一般合成方案。
图9.1显示合成式I的具有1位叔甲酰胺的4-烷基砜前体药物的优选的一般合成方案
图9.1.1显示合成式I的具有酸侧链的4-烷基砜前体药物的一般合成方案
图9.1.2显示合成式I的4-氰基前体药物的一般合成方案
图9.1.3显示合成式I的具有1位叔甲酰胺的4-氰基前体药物的优选的一般合成方案
图9.1.4显示合成式I具有酸侧链的4-氰基前体药物的一般合成方案
图9.2显示合成醇化合物14的方案(图2)
图9.3显示合成醇化合物18的方案(图2)
图9.4显示合成醇化合物301的方案(图2)
图10显示合成磷酸酯10和11和300的方案(图1)
图11显示合成前药22至27的方案(图3)
图12显示合成式II前药的一般方案
图13显示合成醇64和磷酸60的方案
图14显示现有技术前药4(方案1)对本发明的前药10、11、23和300的水性溶解度。表注解:a在包含5%胎牛血清(FCS)的α-最小必需培养基(α-MEM)中测定。b在包含2当量的碳酸氢钠的磷酸盐缓冲液(PBS)中测定。c在pH=4的乳酸盐缓冲液中测定。
图15显示现有技术的前体药物(PR-104A,5至9)在HCT116野生型癌症细胞中对工程化以过表达人两电子还原酶醇醛-酮还原酶1C3(AKR1C3)的HCT116细胞中的IC50(uM)的比较。横跨5至9系列的进展与AKR1C3代谢诱导的细胞毒性相关损失一致,与该低细胞密度试验中PR-104相当,使得前体药物9好像是AKR1C3-阴性的。
图16显示用于NADPH辅助因子损失速度的重组AKR1C3代谢试验。所有的前体药物5至9是对于AKR1C3比PR-104A是更好的底物。
图17显示PR-104A、前体药物7和前体药物9在HCT116野生型多细胞层(MCLs)对HCT116MCLs中的克隆细胞杀伤,其中细胞被工程化,以过表达人两电子还原酶醇醛-酮还原酶1C3(AKR1C3)。当细胞在三维结构中生长时,前体药物7和9都能够产生AKR1C3-依赖性细胞剔除。离开产生它们的细胞的细胞毒素代谢分子能够杀伤邻近细胞。在低细胞密度试验中,它们被稀释在试验培养基中,避免生产的细胞遭受细胞毒性,基本上提供AKR1C3假阴性(见图15)。发明人已经发现MCL试验基本上鉴定没有AKR1C3代谢相关细胞毒性的前体药物。
图17.1显示化合物14被55候选物硝基还原酶过表达文库成员的代谢,如通过GFP SOS试验测量。表示的数据是当用20μM化合物14激发6h时,比较未激发的对照,过表达候选物硝基还原酶的大肠杆菌菌株SOS-R4的微板培养物展示的GFP SOS应答的倍数增加(归一化至培养物密度)。数据是2个独立试验的平均数并且误差棒指示±1标准偏差。虚线指示空质粒对照的基线活性,和对应NfsA和NfsB家族成员的数据集如所标记。Inset:表明化合物14的代谢用于选择NfsA_Ec和NfsA_Bs的单个和多突变体变体。以与上面为55候选物硝基还原酶文库描述的那些相同的方式产生数据集。
图17.2显示化合物18被55候选物硝基还原酶过表达文库成员的代谢,如通过GFP SOS试验测量。表示的数据是当用20μM的化合物18激发6h时,比较未激发的对照,过表达候选物硝基还原酶的大肠杆菌菌株SOS-R4的微板培养物展示的GFP SOS应答的倍数增加(归一化至培养物密度)。数据是2个独立试验的平均数并且误差棒指示±1标准偏差。虚线指示空质粒对照的基线活性,和对应NfsA和NfsB家族成员的数据集如所标记。Inset:表明化合物18的代谢用于选择NfsA_Ec和NfsA_Bs的单个和多突变体变体。以与上面为55候选物硝基还原酶文库描述的那些相同的方式产生数据集。
图17.3显示化合物22被55候选物硝基还原酶过表达文库成员的代谢,如通过GFP SOS试验测量。表示的数据是当用50μM的化合物22激发6h时,比较未激发的对照,过表达候选物硝基还原酶的大肠杆菌菌株SOS-R4的微板培养物展示的GFP SOS应答的倍数增加(归一化至培养物密度)。数据是2个独立试验的平均数并且误差棒指示±1标准偏差。虚线指示空质粒对照的基线活性,和对应NfsA和NfsB家族成员的数据集如所标记。Inset:表明化合物22的代谢用于选择NfsA_Ec和NfsA_Bs的单个和多突变体变体。以与上面为55候选物硝基还原酶文库描述的那些相同的方式产生数据集。
图17.4显示化合物23被55候选物硝基还原酶过表达文库成员的代谢,如通过GFP SOS试验测量。表示的数据是当用60μM的化合物23激发6h时,比较未激发的对照,过表达候选物硝基还原酶的大肠杆菌菌株SOS-R4的微板培养物展示的GFP SOS应答的倍数增加(归一化至培养物密度)。数据是2个独立试验的平均数并且误差棒指示±1标准偏差。虚线指示空质粒对照的基线活性,和对应NfsA和NfsB家族成员的数据集如所标记。Inset:表明化合物23的代谢用于选择NfsA_Ec和NfsA_Bs的单个和多突变体变体。以与上面为55候选物硝基还原酶文库描述的那些相同的方式产生数据集。
图17.5显示化合物24被55候选物硝基还原酶过表达文库成员的代谢,如通过GFP SOS试验测量。表示的数据是当用60μM的化合物24激发6h时,比较未激发的对照,过表达候选物硝基还原酶的大肠杆菌菌株SOS-R4的微板培养物展示的GFP SOS应答的倍数增加(归一化至培养物密度)。数据是2个独立试验的平均数并且误差棒指示±1标准偏差。虚线指示空质粒对照的基线活性,和对应NfsA和NfsB家族成员的数据集如所标记。Inset:表明化合物24的代谢用于选择NfsA_Ec和NfsA_Bs的单个和多突变体变体。以与上面为55候选物硝基还原酶文库描述的那些相同的方式产生数据集。
图17.6显示化合物25被55候选物硝基还原酶过表达文库成员的代谢,如通过GFP SOS试验测量。表示的数据是当用60μM的化合物25激发6h时,比较未激发的对照,过表达候选物硝基还原酶的大肠杆菌菌株SOS-R4的微板培养物展示的GFP SOS应答的倍数增加(归一化至培养物密度)。数据是2个独立试验的平均数并且误差棒指示±1标准偏差。虚线指示空质粒对照的基线活性,和对应NfsA和NfsB家族成员的数据集如所标记。Inset:表明化合物25的代谢用于选择NfsA_Ec和NfsA_Bs的单个和多突变体变体。以与上面为55候选物硝基还原酶文库描述的那些相同的方式产生数据集。
图17.7显示化合物26被55候选物硝基还原酶过表达文库成员的代谢,如通过GFP SOS试验测量。表示的数据是当用60μM的化合物26激发6h时,比较未激发的对照,过表达候选物硝基还原酶的大肠杆菌菌株SOS-R4的微板培养物展示的GFP SOS应答的倍数增加(归一化至培养物密度)。数据是2个独立试验的平均数并且误差棒指示±1标准偏差。虚线指示空质粒对照的基线活性,和对应NfsA和NfsB家族成员的数据集如所标记。Inset:表明化合物26的代谢用于选择NfsA_Ec和NfsA_Bs的单个和多突变体变体。以与上面为55候选物硝基还原酶文库描述的那些相同的方式产生数据集。
图17.8显示化合物27被55候选物硝基还原酶过表达文库成员的代谢,如通过GFP SOS试验测量。表示的数据是当用60μM的化合物27激发6h时,比较未激发的对照,过表达候选物硝基还原酶的大肠杆菌菌株SOS-R4的微板培养物展示的GFP SOS应答的倍数增加(归一化至培养物密度)。数据是2个独立试验的平均数并且误差棒指示±1标准偏差。虚线指示空质粒对照的基线活性,和对应NfsA和NfsB家族成员的数据集如所标记。Inset:表明化合物27的代谢用于选择NfsA_Ec和NfsA_Bs的单个和多突变体变体。以与上面为55候选物硝基还原酶文库描述的那些相同的方式产生数据集。
图17.9显示化合物64被55候选物硝基还原酶过表达文库成员的代谢,如通过GFP SOS试验测量。表示的数据是当用10μM的化合物64激发6h时,比较未激发的对照,过表达候选物硝基还原酶的大肠杆菌菌株SOS-R4的微板培养物展示的GFP SOS应答的倍数增加(归一化至培养物密度)。数据是2个独立试验的平均数并且误差棒指示±1标准偏差。虚线指示空质粒对照的基线活性,和对应NfsA和NfsB家族成员的数据集如所标记。Inset:表明化合物64的代谢用于选择NfsA_Ec和NfsA_Bs的单个和多突变体变体。以与上面为55候选物硝基还原酶文库描述的那些相同的方式产生数据集。
图17.10显示用化合物14、18、22、23、24、25、26、27和64对NfsA_Ec的纯化的酶动力学数据。反应包含10mM Tris-C1(pH 7.0)、4%DMSO、0.20mM NADPH和不同的化合物浓度。通过添加酶启动反应并且在分光光度计上400nm下测量吸光度的改变20s,以监测NTR-催化的化合物还原。使用Sigmaplot 10.0(Systat software Inc.)进行非线性回归分析和米氏(Michaelis-Menten)曲线拟合。
图17.11显示UV/V是对每个测试的化合物在400nm下NfsB_Ec催化的还原的相对速度的光谱测量。将化合物14、18、22、23、24、25、26、27和64(600μM)添加至10mM Tris-Cl中的NADPH(200μM),pH 7.0。通过酶添加启动反应(在0.25和5μg/反应之间,在中试实验中经验确定)。速度表示每分钟添加的每mg酶还原的μmol的化合物。数据是至少4独立测量的平均数并且误差棒指示±1标准偏差。
图18显示前体药物PR-104A和本发明的14、18、22、23、24、25、26、27、64在HCT116野生型癌症细胞对工程化以过表达人两电子还原酶醇醛-酮还原酶1C3(AKR1C3)的HCT116细胞中的IC50(uM)。所有好像比PR-104A较不易受AKR1C3介导的细胞毒性的影响。
图18.1显示前体药物PR-104A和本发明的22、23、24、25、26、27在H1299野生型癌症细胞对工程化以过表达人两电子还原酶醇醛-酮还原酶1C3(AKR1C3)的H1299细胞中的IC50(uM)。所有好像比PR-104A较不易受AKR1C3介导的细胞毒性的影响。
图19显示PR-104A比较本发明的前体药物14、18、22、23、24、25、26、27、64在HCT116野生型多细胞层(MCLs)对HCT116MCLs中的克隆细胞杀伤,其中细胞被工程化,以过表达人两电子还原酶醇醛-酮还原酶1C3(AKR1C3)。前体药物14、18、22、23、24、25、26、27和64不造成AKR1C3依赖性细胞毒性。
图19.1显示式1f的化合物比较它们的状态计算的亲脂性,以表明AKR1C3-依赖性细胞毒性。图的注解:a使用训练的ACD Labs(版本8)logP计算器测定。bAKR1C3-依赖性代谢状态,如通过评估前体药物在HCT116野生型多细胞层(MCLs)对HCT116 MCLs中的克隆细胞杀伤测定,其中细胞被工程化,以过表达人两电子还原酶醇醛-酮还原酶1C3(AKR1C3)。
图20显示前体药物PR-104A和本发明的14、18、22、23、24、25、26、27、64在被工程化以过表达细菌两电子还原酶大肠杆菌NfsA的HCT116野生型癌症细胞对HCT116细胞中IC50(uM)。所有的前体药物产生大肠杆菌NfsA介导的细胞毒性。
图20.1显示前体药物PR-104A和本发明的14、18、22、23、24、25、26、27、64在被工程化以过表达细菌两电子还原酶大肠杆菌NfsA的H1299野生型癌症细胞对H1299细胞中的IC50(uM)。所有的前体药物产生大肠杆菌NfsA介导的细胞毒性。
图21显示PR-104A比较本发明的前体药物14、18、22、23、24、25、26、27、64在用3%被工程化以过表达细菌两电子还原酶大肠杆菌NfsA中的细胞接种的HCT116野生型多细胞层(MCLs)对HCT116MCLs的克隆细胞杀伤,以评估旁观者细胞杀伤。所有的前体药物14、18、22、23、24、25、26、27和64表明被3%表达大肠杆菌NfsA的细胞的代谢,旁观者细胞杀伤是97%的非表达相邻细胞。
图22显示分别以1000、422和1330umol/kg的剂量施用单个剂量的前体药物10、22和60的NIH-III小鼠中15%表达大肠杆菌NfsA的HCT116异种移植物(包含85%野生型细胞)的平均肿瘤体积(mm3)。所有的前体药物显示显著的大肠杆菌NfsA介导的抗肿瘤效力。通过参考显示以338umol/kg的人等价剂量的PR-104的平均肿瘤体积。
图22.1显示分别以338、1000、422和1330umol/kg的剂量施用单个剂量的前体药物PR-104,10、22和60,NIH-III小鼠中,对于15%表达大肠杆菌NfsA的HCT116异种移植物(包含85%野生型细胞)作为仅仅媒介处理的肿瘤对照的百分数的至四倍相对肿瘤体积(RTV4)和肿瘤生长延迟(TGD)的中值时间。
图22.2显示分别以750、422、1330和1330umol/kg的剂量施用单个剂量的前体药物10、22,60和11NIH-III小鼠中15%表达大肠杆菌NfsA的H1299异种移植物(包含85%野生型细胞)的平均肿瘤体积(mm3)。所有的前体药物显示显著的大肠杆菌NfsA介导的抗肿瘤效力。通过参考显示以225mg/kg(388umol/kg)的人等价剂量的PR-104的平均肿瘤体积。
图22.3显示,分别施用338、750、422、1330和1330umol/kg剂量的单个剂量的前体药物PR-104,10、22,60和11的NIH-III小鼠中,对于15%表达大肠杆菌NfsA的H1299异种移植物(包含85%野生型细胞)作为仅仅媒介处理的肿瘤对照的百分数的至四倍相对肿瘤体积(RTV4)和肿瘤生长延迟(TGD)中值时间。
图22.4显示施用500umol/kg的前体药物22、23和26的剂量每天两次(即BID),总共1000umol/kg的单个日剂量的NIH-III小鼠中15%表达大肠杆菌NfsA的H1299异种移植物(包含85%野生型细胞)的平均肿瘤体积(mm3)。所有的前体药物显示显著的大肠杆菌NfsA介导的抗肿瘤效力。
图22.5显示以1000umol/kg(500umol/kgBID)的剂量施用单个剂量的前体药物22、23和26NIH-III小鼠中,对于15%表达大肠杆菌NfsA的H1299异种移植物(包含85%野生型细胞)的作为仅仅媒介处理的肿瘤对照的百分数的至四倍相对肿瘤体积(RTV4)和肿瘤生长延迟(TGD)的中值时间。
图23显示前体药物14、22、18和301在HCT116、H460、H1299和SiHa野生型癌症细胞系和HCT116 POR细胞系中的好氧和缺氧IC50(uM),所述HCT116 POR细胞系是已经被工程化以过表达人单电子还原酶细胞色素P450还原酶的HCT116细胞系。在每个例子中将细胞暴露于缺氧导致产生增加细胞毒性的代谢分子的前体药物的选择性代谢,使得缺氧细胞毒性比(HCR)范围是化合物18和301在野生型癌症细胞系中的11至28倍。在HCT116 POR细胞系中观察到化合物14和22更大的HCR,指示增加的前体药物代谢和所以过表达该人单电子还原酶的细胞中的增加的细胞毒性。
图24显示被工程化以过表达人单电子还原酶细胞色素P450还原酶的HCT116肿瘤异种移植物中‘仅仅前体药物’或‘15Gy照射+前体药物’计算的log细胞杀伤(LCK)。未接收照射的小鼠(黑条)或已经接收15Gy照射小鼠(灰条)中PR-104(345μmol/kg)和前体药物300(1330μmol/kg)。前体药物300显示显著的体内缺氧细胞杀伤。
图25显示NIH-III裸鼠的生长皮下的野生型SiHa肿瘤异种移植物中,‘仅仅前体药物’(单个试剂活性)或‘10Gy照射+前体药物’对仅仅媒介对照的计算的log细胞杀伤(LCK)。小鼠以422umol/kg剂量施用化合物22。没有接收照射(黑条)的小鼠中化合物22的LCK或已经接收化合物22(422umol/kg)和10Gy照射的小鼠的LCK(浅灰色条)。仅仅照射的LCK(小鼠仅仅接收媒介而没有化合物22)显示在具有黑色条纹的浅灰色条。前体药物22显示显著的体内缺氧细胞杀伤,如通过未被10Gy的照射杀菌的肿瘤细胞的克隆细胞杀伤所测定。
图26显示在NIH-III裸鼠的皮下生长的野生型SiHa肿瘤异种移植物中‘仅仅前体药物’(单个试剂活性)或‘10Gy照射+前体药物’的计算的log细胞杀伤(LCK)对仅仅媒介对照。小鼠分别以1330、1330和1000umol/kg施用化合物300,11和23。未接收照射的小鼠的测试化合物的LCK(黑条)或已经接收测试化合物和10Gy照射的小鼠(浅灰色条)。仅仅照射的LCK(小鼠仅仅接收媒介而没有测试化合物)显示在具有黑色条纹的浅灰色条。前体药物300、11和23展示显著的体内缺氧细胞杀伤,如通过没有用10Gy的照射杀菌的的肿瘤细胞的克隆细胞杀伤所测定。对于用化合物300和23的4/4小鼠(通过向上指的箭头所表示),‘前体药物加照射’的LCK>4(即在该试验中超出刻度)。对于用化合物11(+表示)处理的1/4小鼠,‘前体药物加照射’的LCK>4(即在该试验中超出刻度)。
发明详述
定义
PR-104是醇PR-104A的磷酸酯前体药物。PR-104也称为2-((2-溴乙基)(2,4-二硝基-6-((2-(膦酰氧基)乙基)氨甲酰基)苯基)氨基)乙基甲磺酸酯。
PR-104A也称为2-((2-溴乙基)(2-((2-羟乙基)氨甲酰基)-4,6-二硝基苯基)氨基)乙基甲磺酸酯
“硝基还原酶”是可催化硝基官能团(-NO2)或醌官能团还原的酶。如本文提及的硝基还原酶可以是内源性的或外源性的。
“前体药物(Prodrug)”是当活化时转化成活性细胞毒素代谢物的活性化合物。优选地活化通过还原硝基基团发生在局部肿瘤微环境中的表达硝基还原酶的靶细胞中。前体药物可通过不依赖于组织氧浓度的细菌硝基还原酶的还原来活化或通过缺少氧的组织(缺氧组织)中的人硝基还原酶来活化。
提及前体药物,“活化″或″代谢″指与酶接触之后前体药物可经历的催化还原过程。前体药物可被活化/代谢,以产生可选的化合物,比如细胞毒素代谢分子,其可具有用于治疗性应用的有益活性。
“剔除(Ablation)”以其最宽的背景考虑,并且也意思是完全终止被剔除靶的功能,也旨在包括任何程度的抑制靶的功能,其中靶包括但不限于细胞。
“细胞”指生物亚单位,其专门进行具体的一种或多种功能。为了本发明的目的,如本文所定义,术语“细胞”也包括其中发现细胞的培养基。例如这可指肿瘤或细胞基质的缺氧区域,其体内或体外细胞。
“内源性的”-在生物体、组织或细胞中天然存在的,起源的或产生的。例如哺乳动物中的内源性酶是哺乳类的细胞中天然存在的酶。
“外源性的”-在生物体、组织或细胞的外部起源或产生的。例如哺乳动物中的外源性酶是哺乳类的细胞中不存在的外源酶。例如细菌酶可已经通过基因操作来引入。
本文提及“缺氧”指组织中氧的浓度显著低于健康的灌注组织中氧的正常生理学浓度,尤其氧含量小于约1%(百万分之10,000的氧;7.6mmHg)。
“旁观者效应(Bystander effect)”-该效应通过用细胞毒素前体药物代谢物治疗靶细胞激发并且指局部微环境中细胞或组织对靶细胞的二次剔除作用。不希望被理论限制,认为旁观者效应通过细胞毒素前体药物代谢分子(活化前体药物)从产生的位点扩散以影响与靶细胞分开的未修饰的细胞造成。
以其最宽的背景理解“治疗”。术语不必暗示只有完全恢复受试者才被治疗。因此,“治疗”宽泛地包括,例如,预防、改善或控制疾病的一个或多个症状、一个或多个症状的严重程度和预防或以其他方式减少发展二次并发症的风险。
“预防”疾病不应认为暗示疾病发展被完全预防,其包括疾病发展的延迟。
“硝基苯甲酰胺氮芥”指具有用硝基、甲酰胺和苯胺氮芥官能团取代的苯环的任何化合物。
“硝基苯甲酰胺氮芥醇”指具有用硝基、甲酰胺和苯胺氮芥官能团苯环的任何化合物,其中甲酰胺取代基进一步包含醇部分。
“AKR1C3酶”指人酶醇醛-酮还原酶1C3。醇醛-酮还原酶(AKRs)是细胞溶质酶的超家族,其参与将醛和酮从各种内源和外源性底物分别还原成它们相应的伯醇和仲醇(Jez等,1997)。AKR需要存在辅因子NADPH以便催化羰基基团的还原(Schlegel等,1998)。人AKR归类为三个家族-AKR1、AKR6和AKR7-其AKR1根据结构和功能被最佳表征(Penning和Drury,2007)。AKR1C亚家族包括AKR1C1-4,所有的四个酶具有羟基类固醇脱氢酶(HSD)活性(Penning等,2000)。编码AKR1C1-4的基因具有大于86%的氨基酸序列一致性,并且显示底物的不同以及位点代谢的区域专一性(Penning和Byrns,2009)。AKR1C3是负责人中还原前列腺素D2(PGD2)的酶。
本文提及“基本上抗AKR1C3酶代谢”指当与容易被人AKR1C3代谢的化合物比较时,通过人AKR1C3酶展示非常低或基本上零程度代谢的化合物。AKR1C3代谢可通过孵育测试化合物和NADPH辅助因子以及重组AKR1C3蛋白质和分析NADPH辅助因子的损失来测定,其中辅助因子的损失指示化合物的酶代谢。被AKR1C3代谢的本发明的前体药物表明被工程化以过表达AKR1C3的细胞的多细胞层中相对于野生型同基因的细胞的多细胞层增加的克隆细胞杀伤,而基本上抗AKR1C3酶代谢的本发明的化合物表明不能在被工程化以过表达AKR1C3的细胞的多细胞层中相对于野生型同基因的细胞的多细胞层提供增加克隆细胞杀伤。
“药学上可接受的”意思是其用于制备药学组合物一般是安全的、无毒的,并且既不是生物上不期望的也不是以其他方式非期望的并且包括其对于兽医以及人药学用途是可接受的。
化合物“药学上可接受的盐”意思是如本文定义的药学上可接受的盐,并且具有母化合物期望的药理学活性。这种盐包括:
由无机酸比如盐酸、氢溴酸、硫酸、硝酸、磷酸等;或由有机酸比如乙酸、甲磺酸、马来酸、酒石酸柠檬酸等形成的酸加成盐;或
当母化合物中存在的酸性质子被金属离子,例如碱金属离子、碱土离子或铝离子替换时;或与有机或无机碱配位时形成的盐。可接受的有机碱包括乙醇胺、二乙醇胺、N-甲基葡糖胺、三乙醇胺等。可接受的无机碱包括氢氧化铝、氢氧化钙、氢氧化钾、碳酸钠和氢氧化钠。
本文提及“坏死”是死细胞的区域。这些通常见于肿瘤,原因是细胞或组织外因素造成的细胞损伤和永久的细胞死亡,比如来自养分和氧不足供给的创伤。
术语“施用的(administrated)”、“施用(administration)”等,当提及施用化合物至靶细胞时,旨在包括引入的所有方法并且不旨在限于直接施用至肿瘤细胞的位点。术语旨在包括引入化合物至靶细胞,例如使用GDEPT、VDEPT、CDEPT或ADEPT的间接方法。
短语“治疗有效量的”旨在意思是具有引起疗效可能的化合物的量。在前体药物的情况下,本领域是技术人员理解这仅仅在活化/代谢该前体药物之后引起疗效。
本文结合旁观者效应提及“治疗靠近”意思是足够靠近能够代谢/活化前体药物的靶细胞的细胞,使得细胞接收治疗有效浓度的活性/细胞毒素前体药物代谢分子。不希望被理论限制,这通常在靶细胞的1至10个细胞直径内。
下述是概括给出的本发明的说明书,包括其优选的实施方式。从本文下面标题为“实施例”的内容中进一步阐释本发明,其提供支持本发明的实验数据,本发明各个方面的具体实施例,和进行本发明的方式。
本发明广泛涉及新类型的化合物,其具有作为用于癌症疗法和相关方法的试剂或药物的具体应用。尤其,本发明提供了具体类型的硝基苯甲酰胺氮芥、硝基苯甲酰胺氮芥醇和它们相应的磷酸酯,用作靶向细胞毒素试剂或生物还原性前体药物。
现有技术提示AKR1C3活化的消除可在相关类型的二硝基苯甲酰胺氮芥前体药物中实现(Patterson等,WO/2010/044685A1)。在同基因的HCT116细胞系总研究类似的N-烷基甲酰胺系列5至9(方案3),在常规的二维低细胞密度IC50试验中比较野生型或过表达AKR1C3的细胞中化合物的细胞毒性。数据指示增加的N-烷基延伸(H至甲基至乙基至异丙基至丙基)使在需氧条件下AKR1C3依赖性细胞灵敏度消失。所以,得出结论前体药物8和9不是AKR1C3的底物。
方案3
但是,发明人最近测定低细胞密度IC50试验可产生‘假阴性’。随着前体药物系列5至9变得更加亲脂,发明人发现AKR1C3形成的代谢物从细胞生产中丢失进入IC50试验的基本上无限稀释的细胞培养基。该丢失避免细胞遭受细胞毒素损伤,使得最亲脂化合物8和9好像对于AKR1C3依赖性细胞灵敏度是阴性的(图15)。因此,发明人通过重组AKR1C3代谢研究确定整个系列5至9容易被AKR1C3代谢(图16)。
发明人已经显示使AKR1C3阳性细胞在三维层中生长并且然后将多细胞层(MCL)暴露于前体药物5至9与AKR1C3阴性对照MCL实验相比产生广泛的克隆细胞杀伤(图17)。这里,一个细胞中产生的代谢物丢失至邻近细胞,其中提取其细胞毒性。所以,发明人已经发现该高细胞密度MCL筛选提供了从二维体外IC50筛选检测‘假阴性’的方法并且表明其可用于设计真实的AKR1C3-阴性前体药物,比如本发明的那些。
所以WO/2010/044685A1的内容不表示任何形式的硝基苯甲酰胺氮芥、硝基苯甲酰胺氮芥醇或它们相应的磷酸酯可抗酶AKR1C3的代谢。所以,令人吃惊地本发明主题的前体药物类型展示这种对AKR1C3代谢的抗性。
发明人已经鉴定了当与已知的化合物比较时显示被人酶AKR1C3减少的或零代谢的化合物(图18和19)。新的化合物的优势是,它们被缺氧肿瘤区域或表达外源性硝基还原酶的细胞选择性代谢而不是被天然存在于其他组织比如骨髓中的人AKR1C3代谢。该选择性可期望当施用至患者时减少化合物的副作用。选择性也可减少需要的治疗有效的剂量,其优势包括降低成本和潜在的副作用。
先前已知的化合物必须以洁净的DMSO或DMSO/聚乙二醇/水施用(Atwell等,J.Med.Chem.,2007,50,1197-1212),其导致最大耐受剂量的很大变化。本发明化合物的优势是它们在水中的溶解度。这具有溶解化合物用于有效制备本发明的组合物并且确保能够药物代谢动力学计算与剂量和待更精确测量的其他参数相关的优势。增加溶解度也提供更有效施用和帮助有效运输前体药物至体内活化的位点。
在具体的实施方式中,当在包含2当量的碳酸氢钠的磷酸盐缓冲液(PBS)中测定时,第一或第二方面的化合物的溶解度大于约95mM或当在pH=4的乳酸盐缓冲液中测定时,大于10mM。总体上新类型的代表化合物10、11、23和300的溶解度(图14),与化合物4的溶解度0.068相当(当在包含5%胎牛血清(FCS)的α-最小必需培养基(α-MEM)中测定时)。对于待用于该背景的药物大于10mM的溶解度是足够可溶的。所以,本发明的化合物对于用作前体药物展示令人吃惊地适当的溶解度特征。
通过本发明化合物令人吃惊的高溶解度实施的本发明进一步的实施方式是包含本发明化合物的可溶性组合物。这种组合物用于细胞剔除或用于治疗癌症和其他过度增生病况。
本发明的化合物包括硝基苯甲酰胺氮芥、硝基苯甲酰胺氮芥醇和它们相应的磷酸酯。硝基苯甲酰胺氮芥以它们的硝基形式是相对无活性的,但是当还原时转化成一系列活性(细胞毒素)化合物,其可用于细胞剔除,例如肿瘤细胞的剔除。
在具体的实施方式中,本发明提供了细胞剔除的方法,其包括使用本发明的化合物。在具体的实施方式中,化合物是能够通过接触a)至少一种硝基还原酶,和/或b)低氧(缺氧)环境来活化的前体药物。
发明人也已经表明可用于选择化合物的有效方法,所述化合物基本上抗AKR1C3酶代谢(如上定义的)和该化合物在细胞剔除方法中的用途。具体而言,图16显示被AKR1C3代谢的现有技术的化合物,图17显示这些AKR1C3代谢的化合物与野生型细胞相比较,如何在过表达AKR1C3的MCL中产生增加克隆细胞杀伤和图19显示必须抗AKR1C3代谢的本发明的化合物,因为它们不能提供被工程化以过表达AKR1C3细胞的多细胞层相对于野生型同基因的细胞的多细胞层的增加的克隆细胞杀伤。
在进一步的实施方式中,本发明的化合物或其混合物以有效量施用至受试者,以剔除细胞,其中所述细胞表达至少一种硝基还原酶。
本发明的化合物能够渗透肿瘤组织并且通过接触硝基还原酶(图20-22)或通过接触比如肿瘤中出现的缺氧环境选择性还原成活性(细胞毒素)形式。该活性形式能够剔除靶细胞并且所以尤其用于治疗癌症和其他过度增生不适。在具体的实施方式中,硝基还原酶由大肠杆菌的nfsB和/或nfsA基因编码或由其他细菌物种的直向同源基因编码。在可选的实施方式中,硝基还原酶由突变体硝基还原酶编码。本发明提供的化合物可结合使得外源性硝基还原酶在肿瘤中或治疗上接近肿瘤表达的疗法施用至受试者。
在存在人实体瘤中出现的病理学低氧的情况下,能够发生净还原成羟胺和胺细胞毒素代谢分子,提供肿瘤选择性细胞剔除。除了被硝基还原酶代谢,本发明的化合物也在可出现在肿瘤区域中的缺氧区域代谢(图23-25)。因此,在进一步的具体实施方式中,本发明提供了细胞剔除的方法,其包括本发明的化合物或其混合物以有效量施用至剔除细胞的步骤,其中所述细胞出现在缺氧区域中,或治疗上接近缺氧区域。
所以,本发明提供了治疗癌症或过度增生病况的方法,其中本发明的化合物或其混合物以治疗有效量施用至受试者的肿瘤细胞,或治疗上接近肿瘤细胞。
本发明的这种化合物也用于制备剔除细胞的组合物,或用于治疗癌症或过度增生病况的组合物。在具体的实施方式中,化合物是能够通过接触a)至少一种硝基还原酶,和/或b)低氧(缺氧)环境来活化的前体药物。
发明人也已经出人意料地发现本发明的化合物,当结合照射治疗施用至细胞尤其有效剔除细胞(图26)。因此,设想本发明提供了癌症治疗的方法,其并入施用本发明的前体药物和照射肿瘤细胞。照射步骤可在施用前体药物化合物之前、同时或之后进行。
一旦代谢,然后前体药物的活性形式——细胞毒素代谢物能够通过旁观者效应剔除硝基还原酶天然细胞。在三维细胞培养模型测定通过旁观者效应剔除细胞的该能力。该能力尤其用于剔除肿瘤中围绕表达硝基还原酶的细胞的细胞。
发明人先前已经克隆和组装了来自20个细菌物种,代表12个不同酶家族的55个硝基还原酶候选物的系统发生学上多样文库。因为它们共同代谢硝基咪唑成像探针(生物成像)和生物还原性前体药物(生物疗法,生物控制)的能力而筛选了这些细菌硝基还原酶,并且NfsA和NfsB家族已经被鉴定为特别令人感兴趣。硝基还原酶可以是WO/2012/008860中描述的硝基还原酶,其包括突变体硝基还原酶和其中描述的硝基还原酶的功能上等同的变体。
发明人也已经开发了新的筛选方法,以用靶前体药物量化候选物硝基还原酶的活性。使用的筛选方法的进一步的细节和在细菌中NTR筛选前体药物的结果以及代谢试验的NfsA动力学和NfsB速度提供在实施例3和图17.1至17.11中。结果显示测试的化合物有效地被来自许多不同物种和酶家族的细菌硝基还原酶代谢。所以,本发明的化合物对多种细菌硝基还原酶具有广谱亲和性,并且是多种细菌硝基还原酶的底物,具有治疗性用途的潜能同时保持对被内源人还原酶AKR1C3代谢的抗性。筛选选择的化合物是本发明包括的其他化合物组的代表并且可预期类似的结果。
本发明的化合物大体上由式(I)或式(II)定义,其中式(I)是:
其中W表示Cl、Br、I、OSO2R,
X表示Cl、Br、I、OSO2R,
Y表示H、CN、SO2R,
每个R独立地表示低级C1-6烷基基团,
Z选自式(Ia)的任何自由基或其药学上可接受的盐
其中
R1表示H,或低级C1-6烷基基团,
R2和R3可独立地表示H,或低级C1-6烷基基团;或,
R2和R3可连接在一起形成取代的或未取代的包含5元或6元的杂环,
n表示2至6
*表示与式I附接的点;
和,其中式(II)是:
其中W表示Cl、Br、I、OSO2R,
X表示Cl、Br、I、OSO2R,
每个R独立地表示低级C1-6烷基基团,
Z选自式(IIa)的任何自由基或其药学上可接受的盐
其中
R1表示H,或低级C1-6烷基基团,
n表示2至6
*表示与式II附接的点。
式(I)和(II)的化合物可用于治疗或预防癌症或过度增生病况。如先前描述的治疗方法包括施用式I或II化合物,或其药学上可接受的盐,或其混合物至需要其的受试者的步骤。此外,提供了式I或II的化合物或其混合物在制造用于治疗癌症或过度增生病况的组合物中的用途。
在另一实施方式中,提供了细胞剔除的方法,其包括式I或II的化合物,或其药学上可接受的盐,或其混合物以有效量施用,以剔除细胞的步骤,其中所述细胞表达至少一种硝基还原酶或在缺氧环境中。
在本发明进一步的实施方式中,提供了癌症治疗的方法,其中本发明的化合物或其混合物以治疗有效量施用至受试者中的肿瘤细胞。
优选地,表达至少一种硝基还原酶的细胞是受试者组织中的肿瘤细胞。
优选地,细胞是哺乳类的细胞。
在另一实施方式中,提供了癌症治疗的方法,其中式I或II的化合物,或其药学上可接受的盐,以治疗有效量施用至受试者中的肿瘤细胞。
优选地,施用的治疗有效量在所述受试者的最大耐受剂量的约20%至100%之间。
优选地,施用式I或II的化合物,或其药学上可接受的盐,用于结合至少一种硝基还原酶细胞剔除。
优选地,式I或II的化合物,或其药学上可接受的盐,或其混合物结合下述施用:GDEPT(基因导向的酶前体药物疗法)、VDEPT(病毒导向酶前体药物疗法)、CDEPT(梭菌导向的酶前体药物疗法)或ADEPT(抗体导向的酶前体药物疗法)。
优选地,至少一种硝基还原酶由大肠杆菌的nfsA基因或nfsB基因编码或由其他细菌物种的直向同源基因编码。
优选地,癌症治疗的方法进一步包括在施用式I或II的化合物或其混合物之前、期间或之后,施用一个或多个化疗剂和/或疗法至受试者的步骤。
尽管这些化合物通常用于人受试者的癌症预防或癌症疗法,但是它们可用于靶向其他温血动物受试者,比如其他灵长类,农场动物比如牛,和体育动物和宠物比如马,狗和猫的癌症细胞。
在另一实施方式中,提供了药学组合物,其包含治疗有效量的式(I)或式(II)的化合物或其药学上可接受的盐,或其混合物,和药学上可接受的赋形剂、佐剂、载体、缓冲液或稳定剂。
在一种实施方式中,组合物为片剂、胶囊、粉末或液体的形式。组合物可配制为用于肠胃外施用,优选地通过静脉内输注。
在具体的实施方式中,组合物在水溶液中是可溶的。优选地,当在包含2当量的碳酸氢钠的磷酸盐缓冲液(PBS)中测定时,如在组合物中出现的第一或第二方面的化合物的溶解度大于95mM或当在pH=4的乳酸盐缓冲液中测定时,大于10mM。
前体药物的浓度取决于使用的前体药物的性质和一旦被硝基还原酶/缺氧环境活化实现疗效需要的量。但是,应理解实际施用的化合物的量由医生结合相关的情况,包括待治疗的病况、施用的选择路径、实际施用的化合物、年龄、体重和个体患者的应答、患者症状的严重程度等,和需要的治疗,来确定。在某些实施方式中,组合物包含至少一种本发明的化合物,其形式为其药学上可接受的盐、其水合物,或前述任何溶剂化物。本发明的胺的盐可包括氯化物盐、溴化物盐、甲磺酸盐、甲苯磺酸盐、苹果酸盐。本发明的酸的盐可包括钠盐、钙盐、钾酸盐,其中酸包括磷酸和羧酸。
使用的组合物可包括任何前述的药学上可接受的稀释剂、载体、缓冲液、稳定剂、赋形剂和/或佐剂。稀释剂、载体、缓冲液、稳定剂赋形剂和/或佐剂的选择可取决于期望施用的模式等因素。适当的赋形剂的一些例子包括乳糖、右旋糖、蔗糖、山梨糖醇、甘露醇、淀粉、金合欢树胶、磷酸钙、藻酸盐、黄蓍胶、明胶、硅酸钙、微晶纤维素、聚乙烯吡咯烷酮、纤维素、水、糖浆剂和甲基纤维素。组合物可另外包括润滑剂比如滑石、硬脂酸镁和矿物质油、湿润剂、乳化和悬浮剂、防腐剂,比如甲基-和丙基羟基苯甲酸盐、甜味剂、pH调整剂和缓冲剂、毒性调整剂、调味品剂等。通过采用本领域已知的程序,可配制组合物以便在施用至患者之后提供活性成分的快速、持续或延迟释放。组合物可配制为单位剂量形式,每个剂量包含适合作为人和其他哺乳动物的单位剂量的物理上分开的单位,每个单位包含预定量的计算的活性材料,以产生期望的疗效,结合适当的药学赋形剂、稀释剂、载体和/或佐剂。
可根据Wilson等,2002,Cancer Res.62:1425-1432描述的方法量化旁观者效应,其采用由少量(1%)的NTR-表达‘活化剂’细胞,与大部分(99%)的亲本(野生型)‘靶’细胞混合组成的3D多细胞层(MCL)。可测定用于10%生存(C10)的靶细胞(野生型细胞)的前体药物浓度,所述靶细胞不需要激活剂(T)而生长,和共培养(TC)中的靶以及共培养(AC)中激活剂(表达NTR的细胞)。通过旁观者效应效率测定测量测试前体药物的旁观者效应,所述旁观者效应效率可使用算法((LogC10T-LogC10TC)/(LogC10T-Log C10AC))计算。小于约15%、小于约10%、小于约5%、小于约1%或零的BEE值认为“基本上最小的”,但是大于约50%、约60%、约70%的BEE值认为是“显著的”。
实施例
实施例1-化合物合成的材料和方法
合成一系列硝基苯基氮芥前体药物并且通过HPLC、MS、NMR和元素分析表征。
合成式I的4-烷基砜前体药物的一般合成方案显示在图8中。如本领域技术人员所理解,3,4-二氟苯甲醛(100)与烷烃磺酸酯钠的反应提供了烷基砜(III),其可用亚氯酸钠在包含过氧化氢的磷酸缓冲液中氧化,以产生酸(IV)。这些的硝化提供硝基酸(V),其可直接与二乙醇胺反应,以产生二醇(VI),或首先保护,以产生叔丁基酯(VIII),其随后与二乙醇胺反应,以产生二醇(IX)。亚硫酰基氯介导二醇(VI)的氯化作用并且随后所得酸氯化物中间体与脂肪族胺的反应产生1-甲酰胺二氯氮芥(VII),其可经历卤化锂介导的回流的甲乙酮中卤素交换,以产生式I的化合物。可选地二醇(IX)可通过与适当的烷基磺酰基氯的反应转化成它们双烷烃磺酸酯(X)。双烷烃磺酸酯(X)用三氟乙酸的叔丁基酯脱保护产生酸(XI)。这些与草酰氯在存在氧化镁的情况下反应产生了酰基氯中间体,其可进一步与脂肪族胺反应,以产生双烷烃磺酸酯1-甲酰胺衍生物(XII)。这些可与过多的卤化锂在室温下丙酮中反应,以产生式I对称的氮芥,而与1当量的卤化锂在室温下丙酮中反应产生式I不对称的卤基烷烃磺酸酯氮芥。
合成式I的具有1位叔甲酰胺的4-烷基砜前体药物的优选一般合成方案显示在图9.1中。如本领域技术人员所理解,硝基酸(V)可通过与草酰氯反应转化成酰基氯(XIX)。接着,这些与仲脂肪族胺的反应产生叔酰胺(XX),其可与二乙醇胺反应,以产生二醇(XXI)。二醇(XXI)可接着通过与适当的烷基磺酰基氯反应转化成它们双烷烃磺酸酯(XII)。这些可与过多的卤化锂在室温下丙酮中反应,以产生式I对称的氮芥,而与1当量的卤化锂在室温下丙酮中反应产生式I不对称的卤基烷烃磺酸酯氮芥。
合成式I具有酸侧链的4-烷基砜前体药物的一般合成方案显示在图9.1.1中。如本领域技术人员所理解,亚硫酰基氯介导二醇(VI)的氯化作用并且随后所得酰基氯中间体与具有叔丁基酯保护的酸侧链的脂肪族胺的反应,产生1-甲酰胺二氯氮芥(XXII)。卤化锂介导回流的甲乙酮中卤素交换,随后三氟乙酸介导酯脱保护,然后产生式I具有酸侧链的对称的氮芥化合物。可选地,酸(XI)与草酰氯在存在氧化镁的情况下反应产生酰基氯,其可然后与具有叔丁基酯保护的酸侧链的脂肪族胺反应,以产生1-甲酰胺双烷烃磺酸酯氮芥(XXIII)。这些与过多的卤化锂在室温下丙酮中反应,随后三氟乙酸介导酯脱保护然后产生式I具有酸侧链对称的氮芥的化合物。1-甲酰胺双烷烃磺酸酯氮芥(XXIII)与1当量的卤化锂在室温下丙酮中反应,随后三氟乙酸介导酯脱保护,然后产生式I具有酸侧链的不对称的卤基烷烃磺酸酯氮芥。
合成式I的4-氰基前体药物的一般合成方案显示在图9.1.2中。如本领域技术人员所理解,3,4-二氟苯甲醛(100)与氰化钠的反应产生3-氟-4-氰苯甲醛(371)其可用亚氯酸钠在包含过氧化氢的磷酸缓冲液中氧化,以产生3-氟-4-氰基苯甲酸(372)。硝化和随后三氟乙酸酐介导的所得甲酰胺的脱水和水性碱性启动产生酸(373),其被保护为叔丁基酯(374)。与二乙醇胺的反应产生二醇(375),其可通过与适当的烷基磺酰基氯反应转化成双烷烃磺酸酯(XXIV)。双烷烃磺酸酯(XXIV)用三氟乙酸的叔丁基酯脱保护产生酸(XXV)。这些与草酰氯在存在氧化镁的情况下反应产生酰基氯中间体,其可进一步与脂肪族胺反应,以产生双烷烃磺酸酯1-甲酰胺衍生物(XXVI)。这些可与过多的卤化锂在室温下丙酮中反应,以产生式I对称的氮芥,而与1当量的卤化锂在室温下丙酮中反应产生式I不对称的卤基烷烃磺酸酯氮芥。
合成式I的具有1位叔甲酰胺的4-氰基前体药物的优选一般合成方案显示在图9.1.3中。如本领域技术人员所理解,酸(373)可通过与草酰氯反应转化成酰基氯(376)。这与仲脂肪族胺的反应然后产生叔酰胺(XXVII),其可与二乙醇胺反应,以产生二醇(XXVIII)。二醇(XXVIII)可然通过与适当的烷基磺酰基氯反应转化成它们双烷烃磺酸酯(XXVI)。这些可与过多的卤化锂在室温下丙酮中反应,以产生式I对称的氮芥,而与1当量的卤化锂在室温下丙酮中反应产生式I不对称的卤基烷烃磺酸酯氮芥。
合成式I具有酸侧链的4-氰基前体药物的一般合成方案显示在图9.1.4中。如本领域技术人员所理解,酸(XXV)与草酰氯在存在氧化镁的情况下反应产生酰基氯,其可然后与具有叔丁基酯保护的酸侧链的脂肪族胺反应,以产生1-甲酰胺双烷烃磺酸酯氮芥(XXIX)。这些与过多的卤化锂在室温下丙酮中反应,随后三氟乙酸介导酯脱保护然后产生式I具有酸侧链对称的氮芥的化合物。1-甲酰胺双烷烃磺酸酯氮芥(XXIX)与1当量的卤化锂在室温下丙酮中反应,随后三氟乙酸介导酯脱保护,然后产生式I具有酸侧链的不对称的卤基烷烃磺酸酯氮芥。
合成式II前体药物的一般方案显示在图12中。如本领域技术人员所理解,2-氟-5-硝基苯甲酸(125)与亚硫酰基氯反应和随后所得酰基氯与具有THP保护的醇的脂肪族胺反应产生甲酰胺(XIII)。与二乙醇胺的反应然后产生二醇(XIV),其可通过与适当的烷基磺酰基氯反应转化成它们双烷烃磺酸酯(XV)。双烷烃磺酸酯(XV)用适当的烷基磺酸的THP缩醛脱保护产生醇(XVI)。这些可直接与过多的卤化锂在室温下丙酮中反应,以产生式II对称的氮芥醇,而与1当量的卤化锂在室温下丙酮中反应产生式II不对称的卤基烷烃磺酸酯氮芥醇。式II的这些醇类可然后通过首先使它们与二-叔丁基-N,N-二异丙基亚磷酰胺在存在1H-四唑的情况下反应转化成它们各自的磷酸酯,随后用间氯过氧苯甲酸氧化,以分别产生中间叔丁基磷酸酯XVIII和XVII。然后这些用三氟乙酸在二氯甲烷中脱保护产生式II的磷酸酯。
合成醇化合物14(图2)的方案显示在图9.2中。3,4-二氟苯甲醛(100)与甲基亚磺酸钠的反应产生甲基砜101,其用包含过氧化氢的磷酸缓冲液中的亚氯酸钠氧化,以产生酸102。硝化产生酸103,其直接与二乙醇胺反应,以产生二醇104,或首先被保护,以产生叔丁基酯106,其随后与二乙醇胺反应,以产生二醇107。亚硫酰基氯介导二醇104的氯化作用和随后所得酰基氯中间体与2-(甲基氨基)乙醇的反应产生二氯氮芥111,其经历溴化锂介导回流的甲乙酮中卤素交换,以产生化合物14。可选地,二醇107通过与甲磺酰氯反应转化成双甲磺酸酯108。双甲磺酸酯108用三氟乙酸的的叔丁基酯脱保护产生酸109。这与草酰氯在存在氧化镁的情况下反应产生酰基氯中间体,其进一步与2-(甲基氨基)乙醇反应,以产生双甲磺酸酯1-甲酰胺112。这与过多溴化锂在室温下丙酮中反应产生化合物14。
合成醇化合物18(图2)的方案显示在图9.3中。3,4-二氟苯甲醛(100)与乙基亚磺酸钠反应产生乙基砜113,其用包含过氧化氢的磷酸缓冲液中的亚氯酸钠氧化,以产生酸114。硝化产生酸115,其被保护,以产生叔丁基酯116。随后与二乙醇胺反应产生二醇117,其通过与甲磺酰氯反应转化成双甲磺酸酯118。双甲磺酸酯118用三氟乙酸的叔丁基酯脱保护产生酸119。这与草酰氯在存在氧化镁的情况下反应产生酰基氯中间体,其进一步与2-(甲基氨基)乙醇反应,以产生双甲磺酸酯1-甲酰胺120。这与过多溴化锂在室温下丙酮中反应产生化合物18。
合成醇化合物301(图2)的方案显示在图9.4中。亚硫酰基氯介导二醇104的氯化作用和随后所得酰基氯中间体与1-氨基乙醇的反应产生二氯氮芥303,其经历溴化锂介导回流的甲乙酮中卤素交换,以产生化合物301。可选地,双甲磺酸酯109与草酰氯在存在氧化镁的情况下反应,以产生酰基氯中间体,其进一步与1-氨基乙醇反应,以产生双甲磺酸酯1-甲酰胺304。这与过多溴化锂在室温下丙酮中反应产生化合物301。
合成磷酸酯10和11和300(图1)的方案显示在图10中。醇14、18和301与二叔丁基-N,N-二异丙基亚磷酰胺在存在1H-四唑的情况下反应,随后用间氯过氧苯甲酸氧化分别产生叔丁基磷酸酯122、123和302。这些用二氯甲烷中的三氟乙酸脱保护分别产生磷酸酯10、11和300。
合成前体药物22至27(图3)的方案显示在图11中。双甲磺酸酯109与草酰氯在存在氧化镁的情况下反应产生酰基氯中间体,其进一步与1-甲基哌嗪反应,以产生双甲磺酸酯1-甲酰胺124。这与过多溴化锂在室温下丙酮中反应产生化合物22。亚硫酰基氯介导二醇104的氯化作用和随后所得酰基氯中间体与1-乙基哌嗪和1-异丙基哌嗪的反应分别产生二氯氮芥131和132。然后,溴化锂介导回流的甲乙酮中卤素交换,分别产生化合物23和24。双甲磺酸酯119与草酰氯在存在氧化镁的情况下反应产生酰基氯中间体,其进一步与1-甲基哌嗪、1-乙基哌嗪和1-异丙基哌嗪反应,以分别产生双甲磺酸酯1-甲酰胺133、134和135。这些与过多溴化锂在室温下丙酮中反应分别产生化合物25、26和27。
合成醇64和磷酸60的方案显示在图13中。
2-氟-5-硝基苯甲酸125与亚硫酰基氯反应和随后所得酰基氯与乙醇胺的反应产生酰胺136。随后这与3,4-二氢-2H-吡喃在存在催化对甲苯磺酸的情况下THP保护产生酰胺126。这可通过先前描述的酰基氯与THP保护的乙醇胺反应直接制备。然后,酰胺126与二乙醇胺的反应产生二醇127,其通过与甲磺酰氯反应转化成双甲磺酸酯128。用甲磺酸酸的THP缩醛脱保护产生醇129,其与1当量的溴化锂在室温下丙酮中反应,以产生化合物64。这与二叔丁基-N,N-二异丙基亚磷酰胺在存在1H-四唑的情况下反应,随后用间氯过氧苯甲酸氧化产生叔丁基磷酸酯130,其采用二氯甲烷中的三氟乙酸脱保护,以产生磷酸60。
通过HPLC测定化合物在培养基(+5%胎牛血清)、磷酸缓冲盐水溶液(+碳酸氢钠)或乳酸盐缓冲液(pH=4下)的溶解度和稳定性。
磷酸酯化合物通常展示比它们所源自的醇化合物更优的水性溶解度并且本文用作前体药物。已知,它们在血浆中被血清磷酸酶快速清除以释放醇衍生物。
5-(双(2-溴乙基)氨基)-N-(2-羟乙基)-N-甲基-4-(甲基磺酰基)-2-硝基苯甲酰胺(14)
方法1
3-氟-4-(甲基磺酰基)苯甲醛(101)
3,4-二氟苯甲醛100(10.00g,70.37mmol)用DMSO(200mL)中的甲烷亚磺酸钠(10.06g,98.53mmol)在室温下处理。反应混合物在N2中75℃加热3h然后冷却至室温,并且倾倒入冰水的烧杯。通过过滤收集白色固体,用水冲洗,并且在真空炉中45℃下干燥。通过在二氧化硅凝胶上的快速柱色谱纯化粗品用CH2Cl2/己烷(4∶1)然后洁净的CH2Cl2洗脱至产生3-氟-4-(甲基磺酰基)苯甲醛101(11.83g,83%),为白色粉末。M.p.和1HNMR与期望的产物一致。注:在75℃下主要形成期望的异构体。在高于75℃例如90℃的温度下,单甲基磺酰基与双甲基磺酰基的比是~2.4∶1。
3-氟-4-(甲基磺酰基)苯甲酸(102)
向室温下CH3CN(105mL)中的3-氟-4-(甲基磺酰基)苯甲醛101(11.50g,56.87mmol)的溶液中,添加NaH2PO4.4H2O(1.86g,9.69mmol)的缓冲液溶液和水(39.1mL)中的浓HCl(1.2mL)和然后H2O2(35%,9.7mL,285.21mmol)。将反应混合物冷却至0℃并且逐滴添加水(133mL)中的NaClO2(7.21g,79.72mmol)的溶液。在室温下搅拌5h之后,去除溶剂至一半的体积并且通过过滤收集白色固体。用浓HCl处理滤液并且通过过滤沉淀和收集一些更多的产品。在真空炉中45℃下干燥组合的固体,以产生3-氟-4-(甲基磺酰基)苯甲酸102(12.31g,99%),为白色粉末。M.p.和1HNMR与期望的产物一致。
5-氟-4-(甲基磺酰基)-2-硝基苯甲酸(103)
3-氟-4-(甲基磺酰基)苯甲酸102(13.00g,59.58mmol)溶解在浓H2SO4(93mL)并且在室温下逐滴添加发烟HNO3(18mL)。反应混合物在45℃下加热4h,冷却至室温并且倾倒入冰水的烧杯。通过过滤收集固体,用水冲洗数次,并且在真空炉中45℃下干燥,以产生5-氟-4-(甲基磺酰基)-2-硝基苯甲酸103(12.14g,77%),为浅黄色固体。M.p.和1HNMR与期望的产物一致。
5-(双(2-羟乙基)氨基)-4-(甲基磺酰基)-2-硝基苯甲酸(104)
方法1:
5-氟-4-(甲基磺酰基)-2-硝基苯甲酸103(3.00g,11.40mmol)溶解在DMSO(30mL)中并且用二乙醇胺(3.27mL,34.12mmol)在室温下处理。反应混合物在45℃下加热2h,冷却至室温并且倾倒入冰水的烧杯。用乙酸乙酯/异丙醇(EtOAc/i-PrOH)(4∶1)(3x)提取粗黄色树胶并且用水冲洗结合的有机相(6x),用Na2SO4干燥并且在减压下浓缩(水浴35℃),以产生5-(双(2-羟乙基)氨基)-4-(甲基磺酰基)-2-硝基苯甲酸104(3.64g,92%),为黄色树胶。1HNMR[(CD3)2SO]δ14.07(br,s,1H),8.49(s,1H),7.69(s,1H),4.61(br,s,2H),3.57-3.54(m,4H),3.51-3.48(m,4H),3.46(s,3H).HRMS(APCI)计算值为C12H17N2O8S[M+H]+m/z 349.0705:发现值349.0687。
5-(双(2-羟乙基)氨基)-4-(甲基磺酰基)-2-硝基苯甲酸(104)
方法2:
5-氟-4-(甲基磺酰基)-2-硝基苯甲酸103(7.80g,29.64mmol)溶解在DMSO(25mL)和用二乙醇胺(8.51mL,88.79mmol)处理。反应混合物在室温下搅拌2h然后倾倒入冰冷水性HCl(2M,100mL)的烧杯,用EtOAc/i-PrOH(4∶1)提取(3x),用盐水冲洗,用Na2SO4干燥并且在减压下浓缩,以产生5-(双(2-羟乙基)氨基)-4-(甲基磺酰基)-2-硝基苯甲酸104(8.08g,78%),为黄色粉末。
5-(双(2-氯乙基)氨基)-N-(2-羟乙基)-N-甲基-4-(甲基磺酰基)-2-硝基苯甲酰胺(111)
在回流下加热SOCl2(25mL)和DMF(3滴)中5-(双(2-羟乙基)氨基)-4-(甲基磺酰基)-2-硝基苯甲酸104(1.0g,2.87mmol)的搅拌溶液4h。通过在减压下蒸馏去除过多的SOCl2并且残渣溶解在CH2Cl2(10mL)和THF(6mL)中,冷却至0℃并且用2-(甲基氨基)乙醇(822L,10.25mmol)处理。在0℃下搅拌反应混合物20min然后温热至室温,用水性HCl(0.5M,8mL)酸化并且用EtOAc提取(2x)。用盐水冲洗结合的有机相,用Na2SO4干燥并且在减压下蒸发至干燥。通过在二氧化硅凝胶上的快速柱色谱纯化粗品,用CH2Cl2/MeOH(20∶1)洗脱,以产生5-(双(2-氯乙基)氨基)-N-(2-羟乙基)-N-甲基-4-(甲基磺酰基)-2-硝基苯甲酰胺111(390mg,31%),为阻转异构体的混合物,黄色树胶。1HNMR[(CD3)2SO]δ8.66(s,0.4H),8.64(s,0.6H),7.71(s,0.6H),7.66(s,0.4H),4.83-4.78(2t,J=5.4Hz,1H),3.78(br,s,4H),3.76-3.71(m,4H),3.69-3.64(m,1H),3.55-3.52(m,1H),3.48(s,1.6H),3.47(s,1.4H),3.19(br,s,2H),3.04(s,1.6H),2.85(s,1.4H).HRMS(ESI)计算值为C15H21Cl2N3NaO6S[M+Na]+m/z 464.0407:发现值464.0420。
5-(双(2-溴乙基)氨基)-N-(2-羟乙基)-N-甲基-4-(甲基磺酰基)-2-硝基苯甲酰胺(14)
用LiBr(1.26g,14.51mmol)处理3-甲基-2-丁酮(13mL)中的(5-(双(2-氯乙基)氨基)-N-(2-羟乙基)-N-甲基-4-(甲基磺酰基)-2-硝基苯甲酰胺111(320mg,0.72mmol)溶液并且加热至回流过夜。反应混合物冷却至室温并且在减压下去除溶剂。残渣溶解在EtOAc中并且用水冲洗(3x),用Na2SO4干燥并且在减压下浓缩。将粗混合物再提交至LiBr(2x)并且如上处理。通过二氧化硅凝胶上的快速柱色谱纯化终产物,用CH2Cl2/MeOH(20∶1)洗脱并且进一步从CH2Cl2/iPr2O重结晶,以产生5-(双(2-溴乙基)氨基)-N-(2-羟乙基)-N-甲基-4-(甲基磺酰基)-2-硝基苯甲酰胺14(258mg,67%),为阻转异构体的混合物,浅黄色固体:m.p.138-140℃;1HNMR[(CD3)2SO]δ8.65(s,0.4H),8.64(s,0.6H),7.72(s,0.6H),7.67(s,0.4H),4.82-4.77(2t,J=5.5Hz,1H),3.84-3.77(m,4H),3.69-3.59(m,5H),3.58-3.52(m,1H),3.49(s,1.6H),3.48(s,1.4H),3.19(br,s,2H),3.04(s,1.6H),2.86(s,1.4H).分析计算值C15H21Br2N3O6S.0.2iPr2O:C,35.30;H,4.28;N,7.62%;发现值:C,35.06;H,4.09;N,7.64%。
5-(双(2-溴乙基)氨基)-N-(2-羟乙基)-N-甲基-4-(甲基磺酰基)-2-硝基苯甲酰胺(14)
方法2
叔丁基5-氟-4-(甲基磺酰基)-2-硝基苯酸酯(106)
5-氟-4-(甲基磺酰基)-2-硝基苯甲酸103(8.24g,31.31mmol)在50℃下溶解在CH3CN(48mL)中,冷却至室温并且用叔丁基乙酸酯(48mL)和高氯酸(70%,2.64mL,43.83mmol)处理。反应混合物在室温下搅拌48h并且在减压下去除溶剂(水浴50℃)。残渣从冷室中的MeOH/水再结晶。通过过滤收集产物,以产生叔丁基5-氟-4-(甲基磺酰基)-2-硝基苯酸酯106(5.70g,57%),为浅黄色晶体:m.p.99-101℃;1HNMR(CDCl3)δ8.58(d,J=5.7Hz,1H),7.57(d,J=8.5Hz,1H),3.30(s,3H),1.60(s,9H)。分析计算值C12H14FNO6S:C,45.14;H,4.42;N,4.39%;发现值:C,45.44;H,4.47;N,4.32%.注:用水稀释滤液并且用水性HCl(4M)处理,以通过过滤回收一些收集的未反应的开始材料(3.09g)。
叔丁基5-(双(2-羟乙基)氨基)-4-(甲基磺酰基)-2-硝基苯酸酯(107)
用二乙醇胺(2.79mL,29.12mmol)处理DMSO(15mL)中的叔丁基5-氟-4-(甲基磺酰基)-2-硝基苯酸酯106(6.64g,20.79mmol)溶液。反应混合物在室温下搅拌2h然后倾倒入冰水的烧杯,用二乙醚提取(3x),用Na2SO4干燥并且在减压下浓缩。通过二氧化硅凝胶上的快速柱色谱纯化黄色树胶,用CH2Cl2/MeOH(19∶1)洗脱,以产生叔丁基5-(双(2-羟乙基)氨基)-4-(甲基磺酰基)-2-硝基苯酸酯107(7.35g,87%),为黄色树胶。1HNMR[(CD3)2SO]δ8.50(s,1H),7.63(s,1H),4.64(t,J=5.0Hz,2H),3.58-3.54(m,4H),3.52-3.50(m,4H),3.45(s,3H),1.53(s,9H)。HRMS(ESI)C16H25N2O8S[M+H]+m/z计算值405.1347:发现值405.1326。注:在高于室温,例如40至50℃的温度下形成大量的叔丁基5-(2-((2-羟乙基)氨基)乙氧基)-4-(甲基磺酰基)-2-硝基苯酸酯产物(即O-烷基化与N-烷基化竞争)。
叔丁基5-(双(2-((甲基磺酰基)氧)乙基)氨基)-4-(甲基磺酰基)-2-硝基苯酸酯(108)
向CH2Cl2(290mL)和Et3N(10.28mL,73.75mmol)中的叔丁基5-(双(2-羟乙基)氨基)-4-(甲基磺酰基)-2-硝基苯酸酯107(8.52g,21.07mmol)溶液在0℃下逐滴添加MsCl(4.90mL,63.22mmol)。反应混合物在0℃下搅拌20min然后温热至室温,用CH2Cl2稀释,用水冲洗(3x),用Na2SO4干燥并且在减压下浓缩。通过二氧化硅凝胶上的快速柱色谱纯化残渣,用CH2Cl2/MeOH(19∶1)洗脱,以产生叔丁基5-(双(2-((甲基磺酰基)氧)乙基)氨基)-4-(甲基磺酰基)-2-硝基苯酸酯108(10.60g,90%),为黄色树胶。1HNMR[(CD3)2SO]δ8.52(s,1H),7.83(s,1H),4.36(t,J=5.2Hz,4H),3.77(t,J=5.0Hz,4H),3.43(s,3H),3.14(s,6H),1.53(s,9H)。HRMS(ESI)C18H28N2NaO12S3[M+Na]+m/z计算值583.0674:发现值583.0697。
5-(双(2-((甲基磺酰基)氧)乙基)氨基)-4-(甲基磺酰基)-2-硝基苯甲酸(109)
用TFA(21mL)在5℃下处理CH2Cl2(56mL)中的叔丁基5-(双(2-((甲基磺酰基)氧)乙基)氨基)-4-(甲基磺酰基)-2-硝基苯酸酯108(10.60g,18.91)。反应在室温下搅拌2h,并且在减压下去除溶剂。残渣然后溶解在EtOAc中并且蒸发溶剂至干燥,以去除过多的TFA。黄色残渣溶解在CH2Cl2中并且用iPr2O沉淀,以产生5-(双(2-((甲基磺酰基)氧)乙基)氨基)-4-(甲基磺酰基)-2-硝基苯甲酸109(9.54g,100%),为黄色树胶。1HNMR[(CD3)2SO]δ8.50(s,1H),7.89(s,1H),4.35(t,J=5.1Hz,4H),3.75(t,J=5.2Hz,4H),3.44(s,3H),3.14(s,6H)。HRMS(ESI)计算值为C14H20N2NaO12S3[M+Na]+m/z 527.0062:发现值527.0071。
((5-((2-羟乙基)(甲基)氨甲酰基)-2-(甲基磺酰基)-4-硝基苯基)氮烷二基)双(乙烷-2,1-二基)二甲磺酸酯(112)
用MgO(3.19g,79.09mmol)在室温下处理CH2Cl2(80mL)和CH3CN(20mL)中的5-(双(2-((甲基磺酰基)氧)乙基)氨基)-4-(甲基磺酰基)-2-硝基苯甲酸109(2.66g,5.27mmol)溶液,然后冷却至0℃并且用草酰氯(2.71mL,31.62mmol)和DMF(3滴)处理。在0℃下搅拌反应混合物1h然后温热至室温3h。通过硅藻土的短垫过滤混合物并且在减压下去除溶剂。残渣溶解在CH2Cl2(80mL)和THF(20mL)中,冷却至0℃并且用2-(甲基氨基)乙醇(1.27mL,15.85mmol)处理并且温热至室温20min。用水冲洗混合物(3x),用Na2SO4干燥并且在减压下浓缩。通过二氧化硅凝胶上的快速柱色谱纯化残渣,用CH2Cl2/MeOH(19∶1)洗脱,以产生((5-((2-羟乙基)(甲基)氨甲酰基)-2-(甲基磺酰基)-4-硝基苯基)氮烷二基)双(乙烷-2,1-二基)二甲磺酸酯112(2.20g,74%),为阻转异构体的混合物,黄色树胶。1HNMR[(CD3)2SO]δ8.65(s,0.5H),8.64(s,0.5H),7.72(s,0.5H),7.67(s,0.5),4.84-4.79(2t,J=5.2Hz,1H),4.36-4.34(m,4H),3.79-3.76(m,4H),3.68-3.53(m,2H),3.44(s,3H),3.34-3.29(m,2H),3.14(s,6H),3.04(s,1.6H),2.86(s,1.4H)。HRMS(ESI)计算值为C17H27N3NaO12S3[M+Na]+m/z 584.0647:发现值584.0649。
5-(双(2-溴乙基)氨基)-N-(2-羟乙基)-N-甲基-4-(甲基磺酰基)-2-硝基苯甲酰胺(14)
((5-((2-羟乙基)(甲基)氨甲酰基)-2-(甲基磺酰基)-4-硝基苯基)氮烷二基)双(乙烷-2,1-二基)二甲磺酸酯112(3.70g,6.59mmol)溶解在丙酮(200mL)并且用LiBr(11.44g,131.72mmol)在室温下处理。反应混合物搅拌过夜并且去除溶剂。残渣溶解在EtOAc中并且用水冲洗(2x),用Na2SO4干燥并且在减压下浓缩。通过在二氧化硅凝胶上的快速柱色谱纯化粗品用CH2Cl2/MeOH(20∶1)洗脱,以产生5-(双(2-溴乙基)氨基)-N-(2-羟乙基)-N-甲基-4-(甲基磺酰基)-2-硝基苯甲酰胺14(3.13g,89%),为阻转异构体的混合物,黄色固体。M.p.和1HNMR与先前观察相同。
5-(双(2-溴乙基)氨基)-4-(乙磺酰基)-N-(2-羟乙基)-N-甲基-2-硝基苯甲酰胺(18)
4-(乙磺酰基)-3-氟苯甲醛(113)
用DMSO(230mL)中的乙烷亚磺酸钠(23.00g,198.09mmol)在室温下处理3,4-二-氟苯甲醛100(23.50g,165.38mmol)。反应混合物在N2中75℃下加热4h然后冷却至室温,并且倾倒入冰水的烧杯。通过过滤收集白色沉淀,用水冲洗,并且在真空炉中45℃下干燥。然后从CH2Cl2/iPr2O再结晶固体,以产生4-(乙磺酰基)-3-氟苯甲醛113(28.48g,80%),为白色粉末:m.p.107-110℃;1HNMR(CDCl3)δ10.09(d,J=1.9Hz,1H),8.16(dd,J=6.5Hz,1.4,1H),7.87(dd,J=7.9Hz,1.4,1H),7.75(dd,J=9.4Hz,1.4,1H),3.38(2q,J=7.4Hz,2H),1.33(2t,J=7.5Hz,3H)。分析计算值C9H9FO3S:C,49.99;H,4.20;F,8.79%;发现值:C,50.19;H,4.23;F,8.91%。
4-(乙磺酰基)-3-氟苯甲酸(114)
向室温下CH3CN(280mL)中的4-(乙磺酰基)-3-氟苯甲醛113(30.70g,141.98mmol)的溶液,添加NaH2PO4.4H2O(4.65g,24.21mmol)的缓冲液溶液和水(105mL)中的浓HCl(3.2mL)并且然后H2O2(35%,24.1mL,708.62mmol)。将反应混合物冷却至0℃并且逐滴添加水(350mL)中的NaClO2(17.98g,198.81mmol)的溶液。在室温下搅拌5h之后,在减压下去除溶剂至一半的体积和通过过滤收集白色固体。用浓HCl处理滤液并且通过过滤沉淀和收集一些更多的产品。在真空炉中45℃下干燥组合的固体,以产生4-(乙磺酰基)-3-氟苯甲酸114(32.65g,99%),为白色粉末:m.p.184-186℃;1HNMR(CDCl3)δ8.12-8.07(m,2H),7.98-7.95(m,1H),3.38(q,J=7.4Hz,2H),1.34(t,J=7.4Hz,3H)。分析计算值C9H9FO4S:C,46.55;H,3.91;F,8.18%;发现值:C,46.82;H,3.99;F,8.33%。
4-(乙磺酰基)-5-氟-2-硝基苯甲酸(115)
4-(乙磺酰基)3-氟苯甲酸114(15.65g,67.39mmol)溶解在浓H2SO4(107mL)并且在室温下逐滴添加发烟HNO3(21mL)。反应混合物在45℃下加热4h,冷却至室温并且倾倒入冰水的烧杯。通过过滤收集固体,用水冲洗,并且在真空炉中45℃下干燥,以产生4-(乙磺酰基)-5-氟-2-硝基苯甲酸115(16.63g,89%),为浅黄色固体:m.p.140-143℃;1HNMR[(CD3)2SO]δ14.59(br,s,1H),8.41(d,J=5.8Hz,1H),8.07(d,J=9.3Hz,1H),3.53(q,J=7.4Hz,2H),1.20(t,J=7.4Hz,3H)。HRMS(ESI)计算值为C9H9FNO6S[M+1]+m/z 278.0130:发现值278.0129。
叔丁基4-(乙磺酰基)-5-氟-2-硝基苯酸酯(116)
4-(乙磺酰基)-5-氟-2-硝基苯甲酸115(24.35g,87.83mmol)溶解在叔丁基乙酸酯(150mL)中并且用高氯酸(70%,3.70mL,61.48mmol)处理。在室温下搅拌反应混合物过夜然后进一步用高氯酸(70%,3.70mL,61.48mmol)处理并且搅拌24h。然后在减压下去除溶剂(水浴50℃),并且从冷室中的MeOH/水再结晶残渣。通过过滤收集产物并且进一步重结晶CH2Cl2/iPr2O,以产生叔丁基4-(乙磺酰基)-5-氟-2-硝基苯酸酯116(20.51g,70%),为浅黄色晶体:m.p.105-107℃;1HNMR(CDCl3)δ8.54(d,J=5.7Hz,1H),7.54(d,J=8.5Hz,1H),3.37(2q,J=7.6Hz,2H),1.59(s,9H),1.37(2t,J=7.4Hz,3H)。分析计算值C13H16FNO6S.0.1iPr2O:C,47.58;H,5.05;N,4.08%;发现值:C,47.35;H,4.87;N,4.17%。
叔丁基5-(双(2-羟乙基)氨基)-4-(乙磺酰基)-2-硝基苯酸酯(117)
用二乙醇胺(5.06mL,52.81mmol)处理DMSO(30mL)中的叔丁基4-(乙磺酰基)-5-氟-2-硝基苯酸酯116(13.15g,39.45mmol)溶液。反应混合物在室温下搅拌2h然后倾倒入冰水的烧杯,用二乙醚提取(3x),用Na2SO4干燥并且在减压下浓缩。通过二氧化硅凝胶上的快速柱色谱纯化黄色树胶,用CH2Cl2/MeOH(49∶1)洗脱,以产生叔丁基5-(双(2-羟乙基)氨基)-4-(乙磺酰基)-2-硝基苯酸酯117(12.90g,78%),为黄色树胶。1HNMR[(CD3)2SO]δ8.48(s,1H),7.64(s,1H),4.64(t,J=4.8Hz,2H),3.71(q,J=7.3,2H),3.57-3.49(m,8H),1.53(s,9H),0.10(t,J=7.3Hz,3H)。HRMS(ESI)计算值为C17H27N2O8S[M+H]+m/z 419.1483:发现值419.1483。
叔丁基5-(双(2-((甲基磺酰基)氧)乙基)氨基)-4-(乙磺酰基)-2-硝基苯酸酯(118)
在0℃下向叔丁基5-(双(2-羟乙基)氨基)-4-(乙磺酰基)-2-硝基苯酸酯117(12.90g,30.83mmol)in CH2Cl2(500mL)和Et3N(15.04mL,107.90mmol)的溶液逐滴添加MsCl(7.20mL,92.93mmol)。反应混合物在0℃下搅拌20min然后温热至室温,用CH2Cl2稀释,用水冲洗(3x),用Na2SO4干燥并且在减压下浓缩。通过二氧化硅凝胶上的快速柱色谱纯化残渣,用EtOAc/己烷(4∶1)洗脱,以产生叔丁基5-(双(2-((甲基磺酰基)氧)乙基)氨基)-4-(乙磺酰基)-2-硝基苯酸酯118(14.80g,84%),为黄色粉末。1HNMR[(CD3)2SO]δ8.50(s,1H),7.81(s,1H),4.36(t,J=5.0Hz,4H),3.77(t,J=5.0Hz,4H),3.64(q,J=7.4Hz,2H),3.15(s,6H),1.53(s,9H),1.08(t,J=7.3Hz,3H)。HRMS(ESI)计算值为C19H31N2O12S3[M+H]+m/z 575.1020:发现值575.1034。
5-(双(2-((甲基磺酰基)氧)乙基)氨基)-4-(乙磺酰基)-2-硝基苯甲酸119
用TFA(40mL)在5℃下处理CH2Cl2(80mL)中的叔丁基5-(双(2-((甲基磺酰基)氧)乙基)氨基)-4-(乙磺酰基)-2-硝基苯酸酯118(14.80,25.76)。反应在室温下搅拌2h,并且在减压下去除溶剂。残渣然后溶解在EtOAc中并且蒸发溶剂至干燥,以去除过多的TFA。黄色残渣溶解在CH2Cl2中并且用iPr2O沉淀,以产生5-(双(2-((甲基磺酰基)氧)乙基)氨基)-4-(乙磺酰基)-2-硝基苯甲酸119(13.22g,99%),为黄色树胶。1HNMR[(CD3)2SO]δ8.50(s,1H),7.88(s,1H),4.35(t,J=5.0Hz,4H),3.75(t,J=5.0Hz,4H),3.65(q,J=7.3Hz,2H),3.15(s,6H),1.07(t,J=7.4Hz,3H)。HRMS(ESI)计算值为C15H23N2O12S3[M+H]+m/z 519.0413:发现值519.0408。
((5-((2-羟乙基)(甲基)氨甲酰基)-2-(乙磺酰基)-4-硝基苯基)氮烷二基)双(乙烷-2,1-二基)二甲磺酸酯(120)
用MgO(4.99g,123.81mmol)在室温下处理CH2Cl2(50mL)和CH3CN(5mL)中的5-(双(2-((甲基磺酰基)氧)乙基)氨基)-4-(乙磺酰基)-2-硝基苯甲酸119(3.21g,6.19mmol)溶液,然后冷却至0℃并且用草酰氯(3.19mL,37.14mmol)和DMF(3滴)处理。在0℃下搅拌反应混合物1h然后温热至室温3h。通过硅藻土的短垫过滤混合物并且在减压下去除溶剂。残渣溶解在CH2Cl2(105mL)和THF(25mL)中,冷却至0℃并且用2-(甲基氨基)乙醇(1.66mL,20.64mmol)处理并且温热至室温20min。用水冲洗混合物(3x),用Na2SO4干燥并且在减压下浓缩。通过二氧化硅凝胶上的快速柱色谱纯化残渣,用CH2Cl2/MeOH(19∶1)洗脱,以产生((5-((2-羟乙基)(甲基)氨甲酰基)-2-(乙磺酰基)-4-硝基苯基)氮烷二基)双(乙烷-2,1-二基)二甲磺酸酯120(2.35g,66%),为阻转异构体的混合物,黄色树胶。1HNMR[(CD3)2SO]δ8.64(s,0.3H),8.63(s,0.7H),7.71(s,0.4H),7.65(s,0.6H),4.83-4.78(2t,J=5.2Hz,1H),4.36-4.33(m,4H),3.78-3.75(m,4H),3.69-3.53(m,6H),3.15(s,6H),3.04(s,1.6H),2.86(s,1.4H),1.10(t,J=7.4Hz,3H)。HRMS(ESI)计算值为C18H30N3O12S3[M+H]+m/z 576.0972:发现值576.0986。
5-(双(2-溴乙基)氨基)-N-(2-羟乙基)-N-甲基-4-(乙磺酰基)-2-硝基苯甲酰胺(18)
((5-((2-羟乙基)(甲基)氨甲酰基)-2-(乙磺酰基)-4-硝基苯基)氮烷二基)双(乙烷-2,1-二基)二甲磺酸酯120(2.00g,3.47mmol)溶解在丙酮(50mL)并且用LiBr(6.03g,69.49mmol)在室温下处理。反应混合物搅拌过夜并且去除溶剂。残渣溶解在EtOAc中并且用水冲洗(2x),用Na2SO4干燥并且在减压下浓缩。通过在二氧化硅凝胶上的快速柱色谱纯化粗品,用CH2Cl2/MeOH(20∶1)洗脱,以产生5-(双(2-溴乙基)氨基)-N-(2-羟乙基)-N-甲基-4-(乙磺酰基)-2-硝基苯甲酰胺18(1.81g,96%),为阻转异构体的混合物,黄色树胶。1HNMR[(CD3)2SO]δ8.64(s,0.4H),8.63(s,0.6H),7.70(s,0.6H),7.66(s,0.4H),4.83-4.77(2t,J=5.5Hz,1H),3.84-3.76(m,4H),3.73-3.66(m,3H),3.64-3.58(m,4H),3.55-3.52(m,1H),3.23-3.07(m,2H),3.04(s,1.6H),2.86(s,1.4H),1.10(t,J=7.4Hz,3H)。HRMS(ESI)计算值为C16H23Br2N3NaO6S[M+Na]+m/z 565.9556:发现值565.9567。
5-(双(2-溴乙基)氨基)-N-(2-羟乙基)-4-(甲基磺酰基)-2-硝基苯甲酰胺(301)
方法1
5-(双(2-氯乙基)氨基)-N-(2-羟乙基)-4-(甲基磺酰基)-2-硝基苯甲酰胺(303)
在回流下加热搅拌的SOCl2(12.5mL)和DMF(3滴)中5-(双(2-羟乙基)氨基)-4-(甲基磺酰基)-2-硝基苯甲酸104(490mg,1.41mmol)溶液4h。过多的SOCl2通过在减压下蒸馏去除并且残渣溶解在CH2Cl2(5mL)和THF(3mL)中,冷却至0℃并且用2-氨基乙醇(296μL,4.91mmol)处理。在0℃下搅拌反应混合物20min然后温热至室温,用水性HCl(0.5M,4mL)酸化并且用EtOAc提取(2x)。用盐水冲洗结合的有机相,用Na2SO4干燥并且在减压下蒸发至干燥。通过在二氧化硅凝胶上的快速柱色谱纯化粗品,用CH2Cl2/MeOH(25∶1)洗脱,以产生5-(双(2-氯乙基)氨基)-N-(2-羟乙基)-4-(甲基磺酰基)-2-硝基苯甲酰胺303(300mg,50%),为黄色树胶。1HNMR[(CD3)2SO]δ8.80(t,J=5.7Hz,1H),8.51(s,1H),7.69(s,1H),4.79(t,J=5.4Hz,1H),3.81-3.77(m,4H),3.72-3.69(m,4H),3.55-3.51(m,2H),3.48(s,3H),3.34-3.29(m,2H)。LRMS(APCI)计算值为C14H20Cl2N3O68[M+H]+m/z 429.30:发现值429.00。
5-(双(2-溴乙基)氨基)-N-(2-羟乙基)-4-(甲基磺酰基)-2-硝基苯甲酰胺(301)
用LiBr(1.02g,11.75mmol)处理3-甲基-2-丁酮(10mL)中的5-(双(2-氯乙基)氨基)-N-(2-羟乙基)-4-(甲基磺酰基)-2-硝基苯甲酰胺303(250mg,0.58mmol)溶液并且加热至回流过夜。反应混合物冷却至室温并且在减压下去除溶剂。残渣溶解在EtOAc中并且用水冲洗(3x),用Na2SO4干燥并且在减压下浓缩。将粗混合物再提交至LiBr(2x)并且如上处理。通过二氧化硅凝胶上的快速柱色谱纯化终产物,用CH2Cl2/MeOH(20∶1)洗脱,以产生5-(双(2-溴乙基)氨基)-N-(2-羟乙基)-4-(甲基磺酰基)-2-硝基苯甲酰胺301(250mg,83%),为浅黄色固体。M.p.和1HNMR与先前报道的相同(Denny等,WO2005/042471A1)。
5-(双(2-溴乙基)氨基)-N-(2-羟乙基)-4-(甲基磺酰基)-2-硝基苯甲酰胺(301)
方法2
((5-((2-羟乙基)氨甲酰基)-2-(甲基磺酰基)-4-硝基苯基)氮烷二基)双(乙烷-2,1-二基)二甲磺酸酯(304)
用MgO(4.01g,99.60mmol)在室温下处理CH2Cl2(100mL)和CH3CN(25mL)中的5-(双(2-((甲基磺酰基)氧)乙基)氨基)-4-(甲基磺酰基)-2-硝基苯甲酸109(3.35g,6.64mmol)溶液,然后冷却至0℃并且用(3.42mL,39.84mmol)和DMF(3滴)草酰氯处理。在0℃下搅拌反应混合物1h然后温热至室温3h。通过硅藻土的短垫过滤混合物并且在减压下去除溶剂。残渣溶解在CH2Cl2(100mL)和THF(25mL)中,冷却至0℃并且用乙醇胺(601L,9.96mmol)处理并且温热至室温20min。用水冲洗混合物(3x),用Na2SO4干燥并且在减压下浓缩。通过二氧化硅凝胶上的快速柱色谱纯化残渣,用CH2Cl2/MeOH(19∶1)洗脱,以产生((5-((2-羟乙基)氨甲酰基)-2-(甲基磺酰基)-4-硝基苯基)氮烷二基)双(乙烷-2,1-二基)二甲磺酸酯304(3.40g,94%),为黄色树胶。1HNMR[(CD3)2SO]δ8.71(t,J=5.7Hz,1H),8.51(s,1H),7.73(s,1H),4.77(t,J=5.0Hz,1H),4.35(t,J=5.2Hz,4H),3.72(t,J=5.2Hz,4H),3.55-3.51(m,2H),3.44(s,3H),3.34-3.29(m,2H),3.16(s,6H)。HRMS(ESI)计算值为C16H25N3NaO12S3[M+Na]+m/z 570.0497:发现值570.0493。
5-(双(2-溴乙基)氨基)-N-(2-羟乙基)-4-(甲基磺酰基)-2-硝基苯甲酰胺(301)
((5-((2-羟乙基)氨甲酰基)-2-(甲基磺酰基)-4-硝基苯基)氮烷二基)双(乙烷-2,1-二基)二甲磺酸酯304(3.40g,6.21mmol)溶解在丙酮(180mL)中并且用LiBr(10.78g,124.18mmol)在室温下处理。反应混合物搅拌过夜并且去除溶剂。残渣溶解在EtOAc中并且用水冲洗(2x),用Na2SO4干燥并且在减压下浓缩。通过在二氧化硅凝胶上的快速柱色谱纯化粗品用CH2Cl2/MeOH(20∶1)洗脱并且进一步重结晶CH2Cl2/MeOH(4∶1)和iPr2O,以产生5-(双(2-溴乙基)氨基)-N-(2-羟乙基)-4-(甲基磺酰基)-2-硝基苯甲酰胺28(3.06g,95%),为浅黄色固体。M.p.和1HNMR与先前报道的相同(Denny等,WO2005/042471A1)。
2-(5-(双(2-溴乙基)氨基)-N-甲基-4-(甲基磺酰基)-2-硝基苯甲酰氨基)乙基二氢磷酸酯(10)
2-(5-(双(2-溴乙基)氨基)-N-甲基-4-(甲基磺酰基)-2-硝基苯甲酰氨基)乙基二叔丁基磷酸(122)
用二叔丁基-N,N-二异丙基亚磷酰胺(7.44mL,23.56mmol)在5℃下处理DMF(4.2mL)中的5-(双(2-溴乙基)氨基)-N-(2-羟乙基)-N-甲基-4-(甲基磺酰基)-2-硝基苯甲酰胺14(3.13g,5.89mmol)溶液和CH3CN中的1H-四唑溶液(3%,1.90g,27.10mmol)。反应混合物在室温下搅拌4h,用CH2Cl2(25mL)稀释,冷却至0℃并且逐步添加固体m-CPBA(70%,7.78g,44.18mmol)。混合物温热至室温,额外再搅拌的1h并且在减压下去除溶剂。残渣溶解在EtOAc中,用10%亚硫酸钠溶液(2x)和5%碳酸氢钠溶液(3x)冲洗,用Na2SO4干燥并且在减压下浓缩。通过在二氧化硅凝胶上的快速柱色谱纯化粗品,用CH2Cl2/MeOH(25∶1)洗脱,以产生2-(5-(双(2-溴乙基)氨基)-N-甲基-4-(甲基磺酰基)-2-硝基苯甲酰氨基)乙基二叔丁基磷酸122(3.23g,76%),为阻转异构体的混合物,黄色树胶。1HNMR[(CD3)2SO]δ8.67(s,0.5H),8.66(s,0.5H),7.78(s,0.5H),7.60(s,0.5H),4.15-4.02(m,2H),3.85-3.81(m,4H),3.78-3.66(m,2H),3.64-3.61(m,4H),3.49(s,3H),3.07(s,1.5H),2.89(s,1.5H),1.44(s,10H),1.40(s,8H)。HRMS(ESI)计算值为C23H38Br2N3NaO9PS[M+Na]+m/z 744.0301:发现值744.0325。
2-(5-(双(2-溴乙基)氨基)-N-甲基-4-(甲基磺酰基)-2-硝基苯甲酰氨基)乙基二氢磷酸酯(10)
CH2Cl2(17mL)中的2-(5-(双(2-溴乙基)氨基)-N-甲基-4-(甲基磺酰基)-2-硝基苯甲酰氨基)乙基二叔丁基磷酸122(3.23g,4.46mmol)冷却至5℃并且用TFA(17mL)处理。反应混合物在室温下搅拌1h,并且在减压下去除溶剂。残渣用CH2Cl2/iPr2O磨碎然后溶解在CH3CN中。在减压下去除溶剂(水浴29℃),以产生2-(5-(双(2-溴乙基)氨基)-N-甲基-4-(甲基磺酰基)-2-硝基苯甲酰氨基)乙基二氢磷酸酯10(2.72g,100%),为阻转异构体的混合物,黄色树胶。1HNMR[(CD3)2SO]δ8.66(s,0.5H),8.65(s,0.5H),7.78(s,0.5H),7.63(s,0.5H),4.11-4.06(m,2H),3.84-3.81(m,4H),3.78-3.65(m,2H),3.61-3.58(m,4H),3.46(s,3H),3.04(s,1.5H),2.86(s,1.5H)。HRMS(ESI)计算值为C15H22Br2N3NaO9PS[M+Na]+m/z 631.9065:发现值631.9073。
2-(5-(双(2-溴乙基)氨基)-4-(乙磺酰基)-N-甲基-2-硝基苯甲酰氨基)乙基二氢磷酸酯(11)
2-(5-(双(2-溴乙基)氨基)-N-甲基-4-(乙磺酰基)-2-硝基苯甲酰氨基)乙基二叔丁基磷酸(123)
用二叔丁基-N,N-二异丙基亚磷酰胺(4.16mL,13.20mmol)在5℃下处理DMF(2.0mL)中的5-(双(2-溴乙基)氨基)-N-(2-羟乙基)-N-甲基-4-(乙磺酰基)-2-硝基苯甲酰胺18(1.80g,3.30mmol)溶液和CH3CN中的1H-四唑溶液(3%,1.06g,15.18mmol)。反应混合物在室温下搅拌4h,用CH2Cl2(15mL)稀释,冷却至0℃并且逐步添加固体m-CPBA(70%,4.36g,24.75mmol)。混合物温热至室温,额外再搅拌的1h并且在减压下去除溶剂。残渣溶解在EtOAc中,用10%亚硫酸钠溶液(2x)和5%碳酸氢钠溶液(3x)冲洗,用Na2SO4干燥并且在减压下浓缩。通过在二氧化硅凝胶上的快速柱色谱纯化粗品,用CH2Cl2/MeOH(25∶1)洗脱,以产生2-(5-(双(2-溴乙基)氨基)-N-甲基-4-(乙磺酰基)-2-硝基苯甲酰氨基)乙基二叔丁基磷酸123(2.16g,89%),为阻转异构体的混合物,黄色树胶。1HNMR[(CD3)2SO]δ8.65(s,0.5H),8.64(s,0.5H),7.77(s,0.5H),7.59(s,0.5H),4.14-4.11(m,2H),3.84-3.81(m,5H),3.74-3.66(m,3H),3.63-3.60(m,4H),3.07(s,1.5H),2.89(s,1.5H),1.44(s,10H),1.40(s,8H),1.10(t,J=7.3Hz,3H)。HRMS(ESI)计算值为C24H40Br2N3NaO9PS[M+Na]+m/z 758.0469:发现值758.0440。
2-(5-(双(2-溴乙基)氨基)-4-(乙磺酰基)-N-甲基-2-硝基苯甲酰氨基)乙基二氢磷酸酯(11)
CH2Cl2(25mL)中的(5-(双(2-溴乙基)氨基)-N-甲基-4-(乙磺酰基)-2-硝基苯甲酰氨基)乙基二叔丁基磷酸123(2.16g,2.93mmol)冷却至5℃并且用TFA(5mL)处理。反应混合物在室温下搅拌1h,并且在减压下去除溶剂(水浴29℃)。然后用iPr2O磨碎树胶,以产生(5-(双(2-溴乙基)氨基)-4-(乙磺酰基)-N-甲基-2-硝基苯甲酰氨基)乙基二氢磷酸酯11(1.59g,87%),为阻转异构体的混合物,黄色树胶。1HNMR[(CD3)2SO]δ8.65(s,0.6H),8.64(s,0.4H),7.76(s,0.5H),7.62(s,0.5H),4.10-3.99(m,2H),3.84-3.80(m,4H),3.74-3.68(m,3H),3.63-3.57(m,5H),3.06(s,1.4H),2.89(s,1.6H),1.12-1.08(m,3H)。分析计算值C16H24Br2N3O9PS.(0.25iPr2O+0.1CH2Cl2):C,32.10;H,4.16;N,6.38;P,4.70%;发现值:C,32.36;H,4.09;N,6.13;P,4.34%。
2-(5-(双(2-溴乙基)氨基)-4-(甲基磺酰基)-2-硝基苯甲酰氨基)乙基二氢磷酸酯(300)
2-(5-(双(2-溴乙基)氨基)-4-(甲基磺酰基)-2-硝基苯甲酰氨基)乙基二叔丁基磷酸(302)
用二叔丁基-N,N-二异丙基亚磷酰胺(7.32mL,23.20mmol)在5℃下处理DMF(4.1mL)中的5-(双(2-溴乙基)氨基)-N-(2-羟乙基)-4-(甲基磺酰基)-2-硝基苯甲酰胺301(3.00g,5.80mmol)溶液和CH3CN中的1H-四唑溶液(3%,1.87g,26.68mmol)。反应混合物在室温下搅拌4h,用CH2Cl2(25mL)稀释,冷却至0℃并且逐步添加固体m-CPBA(70%,10.22g,58.00mmol)。混合物温热至室温,额外再搅拌的1h并且在减压下去除溶剂。残渣溶解在EtOAc中,用10%亚硫酸钠溶液(2x)和5%碳酸氢钠溶液(3x)冲洗,用Na2SO4干燥并且在减压下浓缩。通过在二氧化硅凝胶上的快速柱色谱纯化粗品,用CH2Cl2/MeOH(25∶1)洗脱,以产生2-(5-(双(2-溴乙基)氨基)-4-(甲基磺酰基)-2-硝基苯甲酰氨基)乙基二叔丁基磷酸302(2.78g,68%),为黄色树胶。1HNMR[(CD3)2SO]δ8.94(t,J=5.6Hz,1H),8.53(s,1H),7.73(s,1H),4.00-3.96(m,2H),3.77-3.74(m,4H),3.64-3.61(m,4H),3.52-3.48(m,2H),3.50(s,3H),1.43(s,18H)。HRMS(ESI)计算值为C22H36Br2N3NaO9PS[M+Na]+m/z 730.0163:发现值730.0169。
2-(5-(双(2-溴乙基)氨基)-4-(甲基磺酰基)-2-硝基苯甲酰氨基)乙基二氢磷酸酯(300)
CH2Cl2(14mL)中的2-(5-(双(2-溴乙基)氨基)-4-(甲基磺酰基)-2-硝基苯甲酰氨基)乙基二叔丁基磷酸302(2.70g,3.81mmol)冷却至5℃并且用TFA(14mL)处理。反应混合物在室温下搅拌1h,并且在减压下去除溶剂。残渣用CH2Cl2/iPr2O磨碎然后溶解在CH3CN中。在减压下去除溶剂(水浴29℃),以产生2-(5-(双(2-溴乙基)氨基)-4-(甲基磺酰基)-2-硝基苯甲酰氨基)乙基二氢磷酸酯300(2.27g,100%),为黄色树胶。1HNMR[(CD3)2SO]δ8.93(t,J=5.8Hz,1H),8.52(s,1H),7.76(s,1H),3.98-3.93(m,2H),3.77-3.74(m,4H),3.64-3.61(m,4H),3.50-3.45(m,2H),3.50(s,3H)。HRMS(ESI)计算值为C14H20Br2N3NaO9PS[M+Na]+m/z 617.8899:发现值617.8917。
(5-(双(2-溴乙基)氨基)-4-(甲基磺酰基)-2-硝基苯基)(4-甲基哌嗪-1-基)甲酮(22)
((5-(4-甲基哌嗪-1-羰基)-2-(甲基磺酰基)-4-硝基苯基)氮烷二基)双(乙烷-2,1-二基)二甲磺酸酯(124)
用MgO(1.23g,30.52mmol)在室温下处理CH2Cl2(20mL)中的5-(双(2-((甲基磺酰基)氧)乙基)氨基)-4-(甲基磺酰基)-2-硝基苯甲酸109(770mg,1.53mmol)溶液和CH3CN(4mL),然后冷却至0℃并且用草酰氯(786μL,9.16mmol)和DMF(3滴)处理。在0℃下搅拌反应混合物1h然后温热至室温4h。通过硅藻土的短垫过滤混合物并且在减压下去除溶剂。残渣溶解在CH2Cl2(20mL)和THF(20mL)中,冷却至0℃并且用1-甲基哌嗪(459mg,4.58mmol)处理并且温热至室温30min。用水冲洗混合物(3x),用Na2SO4干燥并且在减压下浓缩。通过二氧化硅凝胶上的快速柱色谱纯化残渣,用CH2Cl2/MeOH(19∶1)洗脱,以产生((5-(4-甲基哌嗪-1-羰基)-2-(甲基磺酰基)-4-硝基苯基)氮烷二基)双(乙烷-2,1-二基)二甲磺酸酯124(600mg,67%),为黄色树胶。1HNMR[(CD3)2SO]δ8.65(s,1H),7.68(s,1H),4.34(t,J=5.1Hz,4H),3.79-3.78(m,5H),3.49(br,1H),3.44(s,3H),3.24-3.17(m,2H),3.14(s,6H),2.33(br,3H)2.20(s,3H),2.09(br,1H)。HRMS(ESI)计算值为C19H31N4O11S3[M+H]+m/z 587.1146:发现值587.1148。
(5-(双(2-溴乙基)氨基)-4-(甲基磺酰基)-2-硝基苯基)(4-甲基哌嗪-1-基)甲酮(22)
((5-(4-甲基哌嗪-1-羰基)-2-(甲基磺酰基)-4-硝基苯基)氮烷二基)双(乙烷-2,1-二基)二甲磺酸酯124(1.2g,2.05mmol)溶解在丙酮(40mL)中并且用LiBr(3.55g,40.91mmol)在室温下处理。反应混合物搅拌过夜并且去除溶剂。残渣溶解在EtOAc中并且用水冲洗(2x),用Na2SO4干燥并且在减压下浓缩。通过在二氧化硅凝胶上的快速柱色谱纯化粗品,用CH2Cl2/MeOH(20∶1)洗脱,以产生(5-(双(2-溴乙基)氨基)-4-(甲基磺酰基)-2-硝基苯基)(4-甲基哌嗪-1-基)甲酮22(1.01g,89%),为黄色树胶。1HNMR[(CD3)2SO]δ8.65(s,1H),7.69(s,1H),3.85-3.80(m,4H),3.78-3.69(m,2H),3.62(t,J=6.8Hz,4H),3.58-3.51(m,1H),3.48(s,3H),3.23-3.17(m,2H),2.46-2.29(m,2H),2.21(s,3H),2.13(br,1H)。HRMS(ESI)计算值为C17H24Br2KN4O5S[M+K]+m/z 592.9466:发现值592.9474。
(5-(双(2-溴乙基)氨基)-4-(甲基磺酰基)-2-硝基苯基)(4-乙基哌嗪-1-基)甲酮(23)
(5-(双(2-氯乙基)氨基)-4-(甲基磺酰基)-2-硝基苯基)(4-乙基哌嗪-1-基)甲酮(131)
SOCl2(40mL)和DMF(3滴)中的5-(双(2-羟乙基)氨基)-4-(甲基磺酰基)-2-硝基苯甲酸104(1.62g,4.65mmol)搅拌溶液在回流下加热4h。过多的SOCl2通过在减压下蒸馏去除并且残渣溶解在CH2Cl2(32mL)和THF(32mL)中,冷却至0℃并且用1-乙基哌嗪(1.60g,14.01mmol)处理并且温热至室温30min。用水冲洗混合物(3x),用Na2SO4干燥并且在减压下浓缩。通过二氧化硅凝胶上的快速柱色谱纯化残渣,用CH2Cl2/MeOH(19∶1)洗脱,以产生(5-(双(2-氯乙基)氨基)-4-(甲基磺酰基)-2-硝基苯基)(4-乙基哌嗪-1-基)甲酮131(1.12g,50%),为黄色树胶。1HNMR[(CD3)2SO]68.65(s,1H),7.68(s,1H),3.78-3.77(m,8H),3.69(br,1H),3.58(br,1H),3.47(s,3H),3.17(br,2H),2.42(br,1H),2.39-2.33(m,3H),2.25-1.98(m,1H),1.02-0.98(t,J=7.2Hz,3H)。HRMS(ESI)计算值为C18H27Cl2N4O5S[M+H]+m/z 481.1074:发现值481.1073。
(5-(双(2-溴乙基)氨基)-4-(甲基磺酰基)-2-硝基苯基)(4-乙基哌嗪-1-基)甲酮(23)
用LiBr(16.28g,187.46mmol)处理3-甲基-2-丁酮(200ml)中的(5-(双(2-氯乙基)氨基)-4-(甲基磺酰基)-2-硝基苯基)(4-乙基哌嗪-1-基)甲酮131(4.50g,9.37mmol)溶液并且加热至回流过夜。反应混合物冷却至室温并且在减压下去除溶剂。残渣溶解在EtOAc中并且用水冲洗(3x),用Na2SO4干燥并且在减压下浓缩。将粗混合物再提交至LiBr(2x)并且如上处理。通过二氧化硅凝胶上的快速柱色谱纯化终产物,用CH2Gl2/MeOH(20∶1)洗脱,以产生(5-(双(2-溴乙基)氨基)-4-(甲基磺酰基)-2-硝基苯基)(4-乙基哌嗪-1-基)甲酮23(1.92g,36%),为黄色树胶。1HNMR[(CD3)2SO]δ8.65(s,1H),7.69(s,1H),3.83(q,J=7.4Hz,4H),3.74-3.67(m,1H),3.64-3.54(m,6H),3.48(s,3H),3.23-3.16(m,2H),2.46-2.40(m,1H),2.39-2.32(m,3H),2.25-1.98(m,1H),1.02-0.98(t,J=7.2Hz,3H)。HRMS(ESI)计算值为C18H27Br2N4O5S[M+H]+m/z 569.0063:发现值569.0042。
(5-(双(2-溴乙基)氨基)-4-(甲基磺酰基)-2-硝基苯基)(4-异丙基哌嗪-1-基)甲酮(24)
(5-(双(2-氯乙基)氨基)-4-(甲基磺酰基)-2-硝基苯基)(4-异丙基哌嗪-1-基)甲酮(132)
SOCl2(30mL)和DMF(3滴)中的5-(双(2-羟乙基)氨基)-4-(甲基磺酰基)-2-硝基苯甲酸104(1.09g,3.13mmol)搅拌溶液在回流下加热4h。过多的SOCl2通过在减压下蒸馏去除并且残渣溶解在CH2Cl2(20mL)和THF(20mL)中,冷却至0℃并且用1-异丙基哌嗪(1.20g,9.39mmol)处理并且温热至室温30min。用水冲洗混合物(3x),用Na2SO4干燥并且在减压下浓缩。通过二氧化硅凝胶上的快速柱色谱纯化残渣,用CH2Cl2/MeOH(19∶1)洗脱,以产生(5-(双(2-氯乙基)氨基)-4-(甲基磺酰基)-2-硝基苯基)(4-异丙基哌嗪-1-基)甲酮132(1.06g,69%),为黄色树胶。1HNMR[(CD3)2SO]δ8.65(s,1H),7.68(s,1H),3.78-3.77(m,8H),3.68(br,1H),3.56(br,1H),3.47(s,3H),3.16(br,2H),2.74-2.65(m,1H),2.57(br,2H),2.39(br,1H),2.34-2.26(m,1H),0.99(d,J=6.5Hz,6H)。HRMS(ESI)计算值为C19H29Cl2N4O5S[M+H]+m/z 495.1230:发现值495.1217。
(5-(双(2-溴乙基)氨基)-4-(甲基磺酰基)-2-硝基苯基)(4-异丙基哌嗪-1-基)甲酮(24)
用LiBr(3.73g,42.90mmol)处理3-甲基-2-丁酮(100ml)中的(5-(双(2-氯乙基)氨基)-4-(甲基磺酰基)-2-硝基苯基)(4-异丙基哌嗪-1-基)甲酮132(1.06g,2.15mmol)溶液并且加热至回流过夜。反应混合物冷却至室温并且在减压下去除溶剂。残渣溶解在EtOAc中并且用水冲洗(3x),用Na2SO4干燥并且在减压下浓缩。将粗混合物再提交至LiBr(2x)并且如上处理。通过二氧化硅凝胶上的快速柱色谱纯化终产物,用CH2Cl2/MeOH(20∶1)洗脱,以产生(5-(双(2-溴乙基)氨基)-4-(甲基磺酰基)-2-硝基苯基)(4-异丙基哌嗪-1-基)甲酮24(660mg,53%),为黄色树胶。1HNMR[(CD3)2SO]δ8.65(s,1H),7.69(s,1H),3.83(q,J=7.0Hz,4H),3.67-3.53(m,6H),3.50(s,3H),3.18(br,2H),2.73-2.67(m,1H),2.60-2.52(m,1H),2.47-2.40(m,1H),2.33-2.23(m,1H),2.15-2.06(m,1H),0.97(d,J=6.5Hz,6H)。HRMS(ESI)计算值为C19H29Br2N4O5S[M+H]+m/z 583.0220:发现值583.0203。
(5-(双(2-溴乙基)氨基)-4-(乙磺酰基)2-硝基苯基)(4-甲基哌嗪-1-基)甲酮(25)
((5-(4-甲基哌嗪-1-羰基)-2-(乙磺酰基)-4-硝基苯基)氮烷二基)双(乙烷-2,1-二基)二甲磺酸酯(133)
用MgO(3.59g,89.87mmol)在室温下处理CH2Cl2(60mL)和CH3CN(12mL)中的5-(双(2-((甲基磺酰基)氧)乙基)氨基)-4-(乙磺酰基)-2-硝基苯甲酸119(2.33g,4.49mmol)溶液,然后冷却至0℃并且用草酰氯(2.31mL,26.96mmol)和DMF(3滴)处理。在0℃下搅拌反应混合物1h然后温热至室温4h。通过硅藻土的短垫过滤混合物并且在减压下去除溶剂。残渣溶解在CH2Cl2(60mL)和THF(60mL)中,冷却至0℃并且用1-甲基哌嗪(1.35g,13.48mmol)处理并且温热至室温30min。用水冲洗混合物(3x),用Na2SO4干燥并且在减压下浓缩。通过二氧化硅凝胶上的快速柱色谱纯化残渣,用CH2Cl2/MeOH(19∶1)洗脱,以产生((5-(4-甲基哌嗪-1-羰基)-2-(乙磺酰基)-4-硝基苯基)氮烷二基)双(乙烷-2,1-二基)二甲磺酸酯133(1.78g,66%),为黄色树胶。1HNMR[(CD3)2SO]δ8.63(s,1H),7.67(s,1H),4.36-4.33(m,4H),3.80-3.76(m,6H),3.66-3.60(m,3H),3.48(br,2H),3.22-3.17(m,2H),3.15(s,6H),2.20(s,3H),2.13(br,1H),1.10(t,J=7.4Hz,3H)。HRMS(ESI)计算值为C20H33N4O11S3[M+H]+m/z 601.1302:发现值601.1299。
(5-(双(2-溴乙基)氨基)-4-(乙磺酰基)-2-硝基苯基)(4-甲基哌嗪-1-基)甲酮(25)
((5-(4-甲基哌嗪-1-羰基)-2-(乙磺酰基)-4-硝基苯基)氮烷二基)双(乙烷-2,1-二基)二甲磺酸酯133(1.78g,2.96mmol)溶解在丙酮(70mL)中并且用LiBr(5.15g,59.27mmol)在室温下处理。反应混合物搅拌过夜并且去除溶剂。残渣溶解在EtOAc中并且用水冲洗(2x),用Na2SO4干燥并且在减压下浓缩。通过在二氧化硅凝胶上的快速柱色谱纯化粗品,用CH2Cl2/MeOH(20∶1)洗脱,以产生(5-(双(2-溴乙基)氨基)-4-(乙磺酰基)-2-硝基苯基)(4-甲基哌嗪-1-基)甲酮25(1.44g,85%),为黄色树胶。1HNMR[(CD3)2SO]δ8.63(s,1H),7.68(s,1H),3.85-3.80(m,4H),3.72-3.66(m,3H),3.63-3.59(m,5H),3.19(br,2H),2.46-2.29(m,3H),2.21(s,3H),2.17(br,1H),1.11(t,J=7.4Hz,3H)。HRMS(ESI)计算值为C18H26Br2KN4O5S[M+K]+m/z 606.9622:发现值606.9615。
(5-(双(2-溴乙基)氨基)-4-(乙磺酰基)-2-硝基苯基)(4-乙基哌嗪-1-基)甲酮(26)
((5-(4-乙基哌嗪-1-羰基)-2-(乙磺酰基)-4-硝基苯基)氮烷二基)双(乙烷-2,1-二基)二甲磺酸酯(134)
用MgO(3.02g,75.60mmol)在室温下处理CH2Cl2(50mL)和CH3CN(10mL)中的5-(双(2-((甲基磺酰基)氧)乙基)氨基)-4-(乙磺酰基)-2-硝基苯甲酸119(1.96g,3.78mmol)溶液,然后冷却至0℃并且用草酰氯(1.95mL,22.68mmol)和DMF(3滴)处理。在0℃下搅拌反应混合物1h然后温热至室温4h。通过硅藻土的短垫过滤混合物并且在减压下去除溶剂。残渣溶解在CH2Cl2(50mL)和THF(50mL)中,冷却至0℃并且用1-乙基哌嗪(1.29g,11.34mmol)处理并且温热至室温30min。用水冲洗混合物(3x),用Na2SO4干燥并且在减压下浓缩。通过二氧化硅凝胶上的快速柱色谱纯化残渣,用CH2Cl2/MeOH(19∶1)洗脱,以产生((5-(4-乙基哌嗪-1-羰基)-2-(乙磺酰基)-4-硝基苯基)氮烷二基)双(乙烷-2,1-二基)二甲磺酸酯134(1.14g,49%),为黄色树胶。1HNMR[(CD3)2SO]δ8.63(s,1H),7.67(s,1H),4.34-4.33(m,4H),3.80-3.76(m,5H),3.66-3.60(m,2H),3.49(br,2H),3.22-3.17(m,2H),3.15(s,6H),2.39-2.32(m,4H),2.18(br,1H),1.09(t,J=7.4Hz,3H),1.00(t,J=7.2Hz,3H)。HRMS(ESI)计算值为C21H35N4O11S3[M+H]+m/z 615.1459:发现值615.1464。
(5-(双(2-溴乙基)氨基)-4-(乙磺酰基)-2-硝基苯基)(4-乙基哌嗪-1-基)甲酮(26)
((5-(4-乙基哌嗪-1-羰基)-2-(乙磺酰基)-4-硝基苯基)氮烷二基)双(乙烷-2,1-二基)二甲磺酸酯134(1.14g,1.85mmol)溶解在丙酮(45mL)中并且用LiBr(5.15g,37.09mmol)在室温下处理。反应混合物搅拌过夜并且去除溶剂。残渣溶解在EtOAc中并且用水冲洗(2x),用Na2SO4干燥并且在减压下浓缩。通过在二氧化硅凝胶上的快速柱色谱纯化粗品,用CH2Cl2/MeOH(20∶1)洗脱,以产生(5-(双(2-溴乙基)氨基)-4-(乙磺酰基)-2-硝基苯基)(4-乙基哌嗪-1-基)甲酮26(915mg,85%),为黄色树胶。1HNMR[(CD3)2SO]δ8.63(s,1H),7.68(s,1H),3.84-3.80(m,4H),3.77-3.66(m,4H),3.63-3.59(m,5H),3.19(br,2H),2.42(br,1H),2.39-2.32(m,3H),2.21(br,1H),1.11(t,J=7.4Hz,3H),1.00(t,J=7.2Hz,3H)。HRMS(ESI)计算值为C19H29Br2N4O5S[M+H]+m/z 583.0220:发现值583.0221。
(5-(双(2-溴乙基)氨基)-4-(乙磺酰基)-2-硝基苯基)(4-异丙基哌嗪-1-基)甲酮(27)
((5-(4-异丙基哌嗪-1-羰基)-2-(乙磺酰基)-4-硝基苯基)氮烷二基)双(乙烷-2,1-二基)二甲磺酸酯(135)
用MgO(3.08g,77.14mmol)在室温下处理CH2Cl2(50mL)和CH3CN(12mL)中的5-(双(2-((甲基磺酰基)氧)乙基)氨基)-4-(乙磺酰基)-2-硝基苯甲酸119(2.00g,3.85mmol)溶液,然后冷却至0℃并且用草酰氯(1.99mL,23.14mmol)和DMF(3滴)处理。在0℃下搅拌反应混合物1h然后温热至室温4h。通过硅藻土的短垫过滤混合物并且在减压下去除溶剂。残渣溶解在CH2Cl2(50mL)和THF(50mL)中,冷却至0℃并且用1-异丙基哌嗪(1.48g,11.57mmol)处理并且温热至室温30min。用水冲洗混合物(3x),用Na2SO4干燥并且在减压下浓缩。通过二氧化硅凝胶上的快速柱色谱纯化残渣,用CH2Cl2/MeOH(19∶1)洗脱,以产生((5-(4-异丙基哌嗪-1-羰基)-2-(乙磺酰基)-4-硝基苯基)氮烷二基)双(乙烷-2,1-二基)二甲磺酸酯135(1.21g,49%),为黄色树胶。1HNMR[(CD3)2SO]δ8.63(s,1H),7.67(s,1H),4.34-4.33(m,4H),3.78-3.77(m,5H),3.66-3.60(m,2H),3.53-3.45(m,1H),3.20(br,2H),3.16(s,6H),2.73-2.66(m,1H),2.58(br,1H),2.45-2.25(m,3H),1.09(t,J=7.4Hz,3H),0.97(d,J=6.5Hz,6H)。HRMS(ESI)计算值为C22H37N4O11S3[M+H]+m/z 629.1615:发现值629.1612。
(5-(双(2-溴乙基)氨基)-4-(乙磺酰基)-2-硝基苯基)(4-异丙基哌嗪-1-基)甲酮(27)
((5-(4-异丙基哌嗪-1-羰基)-2-(乙磺酰基)-4-硝基苯基)氮烷二基)双(乙烷-2,1-二基)二甲磺酸酯135(1.16g,2.58mmol)溶解在丙酮(45mL)中并且用LiBr(3.06g,35.22mmol)在室温下处理。反应混合物搅拌过夜并且去除溶剂。残渣溶解在EtOAc中并且用水冲洗(2x),用Na2SO4干燥并且在减压下浓缩。通过在二氧化硅凝胶上的快速柱色谱纯化粗品,用CH2Cl2/MeOH(20∶1)洗脱,以产生(5-(双(2-溴乙基)氨基)-4-(乙磺酰基)-2-硝基苯基)(4-异丙基哌嗪-1-基)甲酮27(934mg,85%),为黄色树胶。1HNMR[(CD3)2SO]δ8.63(s,1H),7.68(s,1H),3.84-3.81(m,4H),3.77-3.66(m,3H),3.63-3.53(m,5H),3.18(br,2H),2.73-2.67(m,1H),2.57(br,2H),2.40(br,1H),2.31(br,1H),1.10(t,J=7.4Hz,3H),0.97(d,J=6.5Hz,6H)。HRMS(ESI)计算值为C20H31Br2N4O5S[M+H]+m/z 597.0376:发现值597.0394。
2-((2-溴乙基)(2-((2-羟乙基)氨甲酰基)-4-硝基苯基)氨基)乙基甲磺酸酯(64)
2-氟-5-硝基-N-(2-((四氢-2H-吡喃-2-基)氧)乙基)苯甲酰胺(126)
方法1
SOCl2(50mL)和DMF(3滴)中的2-氟-5-硝基苯甲酸125(4.56g,24.63mmol)搅拌溶液在回流下加热4h。过多的SOCl2通过在减压下蒸馏去除并且残渣溶解在THF(30mL)中,冷却至-10℃并且用2-((四氢-2H-吡喃-2-基)氧)乙胺(3.93g,27.10mmol)处理并且温热至室温30min。蒸发溶剂并且残渣溶解在EtOAc中,用水和盐水冲洗(3x),用Na2SO4干燥并且在减压下浓缩。通过在二氧化硅凝胶上的快速柱色谱纯化粗品,用EtOAc/己烷(1∶1)洗脱,以产生2-氟-5-硝基-N-(2-((四氢-2H-吡喃-2-基)氧)乙基)苯甲酰胺126(3.17g,41%),为无色油。1HNMR[(CD3)2SO]δ8.67(br,1H),8.42-8.38(m,2H),7.64-7.59(m,1H),4.62(t,J=3.5Hz,1H),3.55-3.41(m,5H),1.79-1.70(m,1H),1.66-1.60(m,1H),1.53-1.41(m,5H)。HRMS(ESI)计算值为C14H17FN2NaO5[M+Na]+m/z 335.1014:发现值335.1014。
方法2
SOCl2(160mL)和DMF(3滴)中2-氟-5-硝基苯甲酸125(15.0g,81.08mmol)的搅拌溶液在回流下加热4h。过多的SOCl2通过在减压下蒸馏去除并且残渣溶解在THF(100mL)中,冷却至-10℃并且用乙醇胺(8.53mL,141.81mmol)处理并且温热至室温30min。蒸发溶剂并且残渣溶解在EtOAc中,用水和盐水冲洗(3x),用Na2SO4干燥并且在减压下浓缩。通过在二氧化硅凝胶上的快速柱色谱纯化粗品,用EtOAc/己烷(2∶1)洗脱,以产生2-氟-N-(2-羟乙基)-5-硝基苯甲酰胺136(17.0g,92%),为无色树胶。1HNMR[(CD3)2SO]δ8.58(br,1H),8.46-8.44(m,1H),8.41-8.31(m,1H),7.60(t,J=9.3Hz,1H),4.79(t,J=5.6Hz,1H),3.55-3.50(m,2H),3.35-3.32(m,2H)。
2-氟-N-(2-羟乙基)-5-硝基苯甲酰胺136(17.0g,74.50mmol)溶解在CH2Cl2(300mL)中并且用催化量的4-甲基苯磺酸(1.28g,7.45mmol)随后3,4-二氢-2H-吡喃(13.59mL,149.01mmol)在室温下处理。反应混合物搅拌过夜,然后用水冲洗(3x),用Na2SO4干燥并且在减压下浓缩。通过在二氧化硅凝胶上的快速柱色谱纯化粗品,用EtOAc/己烷(1∶1)洗脱,以产生2-氟-5-硝基-N-(2-((四氢-2H-吡喃-2-基)氧)乙基)苯甲酰胺126(16.8g,72%),为无色油。1HNMR和HRMS与方法1一致。
2-(双(2-羟乙基)氨基)-5-硝基-N-(2-((四氢-2H-吡喃-2-基)氧)乙基)苯甲酰胺(127)
2-氟-5-硝基-N-(2-((四氢-2H-吡喃-2-基)氧)乙基)苯甲酰胺126(3.17g,10.15mmol)溶解在二恶烷(150mL)并且用Et3N(4.24mL,30.45mmol)和二乙醇胺(3.89mL,40.60mmol)处理。反应混合物加热至55℃过夜,然后冷却至室温,并且蒸发溶剂。残渣溶解在EtOAc中,用水和盐水冲洗(3x),用Na2SO4干燥并且在减压下浓缩。通过在二氧化硅凝胶上的快速柱色谱纯化粗品,用EtOAc/己烷(3∶1)洗脱,以产生2-(双(2-羟乙基)氨基)-5-硝基-N-(2-((四氢-2H-吡喃-2-基)氧)乙基)苯甲酰胺127(3.51g,87%),为黄色树胶。1HNMR[(CD3)2SO]δ8.71(t,J=5.5Hz,1H),8.10-8.06(m,2H),7.17(d,J=9.0Hz,1H),4.73(t,J=5.3Hz,2H),4.62-4.60(m,1H),3.80-3.72(m,2H),3.57-3.49(m,5H),3.47-3.40(m,7H),1.77-1.70(m,1H),1.66-1.61(m,1H),1.50-1.46(m,4H)。
((4-硝基-2-((2-((四氢-2H-吡喃-2-基)氧)乙基)氨甲酰基)苯基)氮烷二基)双(乙烷-2,1-二基)二甲磺酸酯(128)
CH2Cl2(180mL)中的2-(双(2-羟乙基)氨基)-5-硝基-N-(2-((四氢-2H-吡喃-2-基)氧)乙基)苯甲酰胺127(6.01g,15.12mmol)溶液冷却至0℃并且用Et3N(7.38mL,52.93mmol)随后甲磺酰氯(4.40mL,45.36mmol)处理。反应混合物温热至室温30min,用水冲洗(3x),用Na2SO4干燥并且在减压下浓缩。通过二氧化硅凝胶上的快速柱色谱纯化残渣,用CH2Cl2/MeOH(20∶1)洗脱,以产生((4-硝基-2-((2-((四氢-2H-吡喃-2-基)氧)乙基)氨甲酰基)苯基)氮烷二基)双(乙烷-2,1-二基)二甲磺酸酯128(8.04g,96%),为黄色树胶。1HNMR[(CD3)2SO]δ8.73(t,J=5.5Hz,1H),8.15-8.10(m,2H),7.27(d,J=9.2Hz,1H),4.32(t,J=5.3Hz,4H),3.82-3.74(m,6H),3.56-3.40(m,5H),3.13(s,6H),1.76-1.71(m,1H),1.68-1.61(m,1H),1.50-1.47(m,4H)。
((2-((2-羟乙基)氨甲酰基)-4-硝基苯基)氮烷二基)双(乙烷-2,1-二基)二甲磺酸酯(129)
用甲磺酸酸(17.8mL,20.37mmol)处理干燥MeOH(100mL)中的((4-硝基-2-((2-((四氢-2H-吡喃-2-基)氧)乙基)氨甲酰基)苯基)氮烷二基)双(乙烷-2,1-二基)二甲磺酸酯128(3.03g,5.47mmol)。反应混合物在室温下搅拌20min并且蒸发溶剂。残渣溶解在EtOAc中,用水冲洗(3x),用Na2SO4干燥并且在减压下浓缩和通过在二氧化硅凝胶上的快速柱色谱纯化粗品,用CH2Cl2/MeOH(19∶1)洗脱,以产生((2-((2-羟乙基)氨甲酰基)-4-硝基苯基)氮烷二基)双(乙烷-2,1-二基)二甲磺酸酯129(1.92g,75%),为黄色树胶。1HNMR[(CD3)2SO]δ8.73(t,J=5.5Hz,1H),8.15-8.10(m,2H),7.27(d,J=9.2Hz,1H),4.32(t,J=5.3Hz,4H),3.82-3.74(m,6H),3.56-3.40(m,5H),3.13(s,6H),1.76-1.71(m,1H),1.68-1.61(m,1H),1.50-1.47(m,4H)。HRMS(ESI)计算值为C15H23N3NaO10S2[M+Na]+m/z 492.0717:发现值492.0720。
2-((2-溴乙基)(2-((2-羟乙基)氨甲酰基)-4-硝基苯基)氨基)乙基甲磺酸酯(64)
((2-((2-羟乙基)氨甲酰基)-4-硝基苯基)氮烷二基)双(乙烷-2,1-二基)二甲磺酸酯129(8.00g,17.04mmol)溶解在丙酮(240mL)中并且用LiBr(1.48g,17.04mmol)在室温下处理。反应混合物搅拌过夜并且去除溶剂。残渣溶解在EtOAc中并且用水冲洗(2x),用Na2SO4干燥并且在减压下浓缩。通过在二氧化硅凝胶上的快速柱色谱纯化粗品,用CH2Cl2/MeOH(20∶1)洗脱,以产生2-((2-溴乙基)(2-((2-羟乙基)氨甲酰基)-4-硝基苯基)氨基)乙基甲磺酸酯64(3.74g,48%),为黄色树胶。1HNMR[(CD3)2SO]δ8.65(t,J=5.6Hz,1H),8.14-8.09(m,2H),7.22(d,J=9.1Hz,1H),4.75(br,1H),4.31(t,J=5.4Hz,2H),3.80-3.74(m,5H),3.65-3.53(m,5H),3.13(s,3H)。HRMS(ESI)计算值为C14H21BrN3O7S[M+H]+m/z 454.0278:发现值454.0273。
2-((2-溴乙基)(4-硝基-2-((2-(膦酰氧基)乙基)氨甲酰基)苯基)氨基)乙基甲磺酸酯(60)
2-((2-溴乙基)(2-((2-((二-叔丁氧基磷酰基)氧)乙基)氨甲酰基)-4-硝基苯基)氨基)乙基甲磺酸酯(130)
在10℃下用1H-四唑(61.9mL,26.50mmol;3%w/w溶液in MeCN)处理DMF(2mL)中的2-((2-溴乙基)(2-((2-羟乙基)氨甲酰基)-4-硝基苯基)氨基)乙基甲磺酸酯64(2.61g,5.76mmol)搅拌溶液,随后缓慢添加二叔丁基二异丙基氨基磷酸酯(95%,7.68mL,23.05mmol)。混合物在室温下搅拌4h,然后冷却至5℃并且用逐步添加的m-CPBA(70%,7.46g,43.21mmol)处理。在室温下进一步搅拌2h之后,在减压下浓缩反应混合物并且残渣溶解在EtOAc中,用10%水性Na2S2O5、5%水性NaHCO3和水冲洗,然后用Na2SO4干燥并且在减压下浓缩。通过在二氧化硅凝胶上的快速柱色谱纯化粗品,用EtOAc/己烷(1∶1)洗脱,以产生2-((2-溴乙基)(2-((2-((二-叔丁氧基磷酰基)氧)乙基)氨甲酰基)-4-硝基苯基)氨基)乙基甲磺酸酯130(1.45g,39%),为黄色树胶。1HNMR[(CD3)2SO]δ8.84(t,J=5.6Hz,1H),8.13(2d,J=2.8Hz,1H),8.09(d,J=2.8Hz,1H),7.23(d,J=9.3Hz,1H),4.32(t,J=5.4Hz,2H),4.00(q,J=6.3Hz,2H),3.80-3.74(m,4H),3.64(t,J=6.7Hz,2H),3.51(q,J=5.6Hz,2H),3.14(s,3H),1.42(s,18H)。HRMS(ESI)计算值为C22H37BrKN3O10PS[M+K]+m/z 684.0752:发现值684.0740。
2-((2-溴乙基)(4-硝基-2-((2-(膦酰氧基)乙基)氨甲酰基)苯基)氨基)乙基甲磺酸酯(60)
用TFA(10mL)在室温下处理CH2Cl2(30mL)中的2-((2-溴乙基)(2-((2-((二-叔丁氧基磷酰基)氧)乙基)氨甲酰基)-4-硝基苯基)氨基)乙基甲磺酸酯130(1.45g,2.24mmol)搅拌溶液,搅拌1h,然后在减压下浓缩,以去除过多的TFA。所得黄色残渣溶解在EtOAc中并且蒸发至干燥,以产生2-((2-溴乙基)(4-硝基-2-((2-(膦酰氧基)乙基)氨甲酰基)苯基)氨基)乙基甲磺酸酯60(1.20g,100%),为黄色树胶。1HNMR[(CD3)2SO]δ8.86(t,J=5.6Hz,1H),8.15-8.11(m,2H),7.24(d,J=9Hz,1H),4.32(t,J=5.4Hz,2H),4.07-4.02(m,2H),3.79-3.74(m,4H),3.65(t,J=6.7Hz,2H),3.51(q,J=5.5Hz,2H),3.13(s,3H)。HRMS(ESI)计算值为C14H21BrKN3O10PS[M+K]+m/z 571.9500:发现值571.9451。
实施例2-前体药物细胞毒性筛选
化合物经受测试,包括低细胞密度细胞毒性试验。细胞(500个细胞/孔)接种在96孔板中并且暴露于前体药物4h,冲洗,然后生长另外的5天。采用SRB试验,以测定IC50值(抑制增殖50%需要的前体药物的浓度)。头对头评估亲本HCT116细胞和表达来自大肠埃希杆菌示的例性硝基还原酶基因nfsA的细胞。头对头评估亲本H1299细胞和表达来自大肠埃希杆菌的示例性硝基还原酶基因nfsA的细胞。头对头评估亲本HCT116细胞和表达人AKR1C3的细胞。头对头评估亲本H1299细胞和表达人AKR1C3的细胞。在这些试验测试的变型中,评估化合物它们的产生低氧选择性细胞杀伤的能力。这里使用野生型HCT116、H460、H1299和SiH细胞,除了被工程化以过表达细胞色素P450还原酶(HCT116POR),人单电子硝基还原酶的HCT116细胞。如上在好氧条件下进行低细胞密度细胞毒性试验,并且比较其中细胞在前体药物暴露期间显示4h缺氧的实验,然后冲洗不含前体药物并且接着如上在好氧条件下生长另外的5天。
平行进行三维高细胞密度多细胞层(MCL)克隆试验。1x106个100%HCT-116WT或包含97%WT和少数3%HCT-116NTR细胞群体的细胞接种在包被胶原的Teflon微孔膜中并且生长3天。然后将MCL暴露于前体药物5h。处理之后,MCL被酶分解,稀释在新鲜培养基中并且铺平板,以确定克隆生存。为了区分克隆活化剂(NTR+)和靶(NTR-)克隆,将细胞铺平板在非选择性培养基(全细胞)和包含1μM嘌呤霉素(活化剂细胞)的培养基。克隆生长10天,然后染色。计数包含>50个细胞的克隆。该试验的变型也用于评估AKR1C3-依赖性细胞毒性的证据。这里,接种100%HCT-116WT或100%HCT-116AKR1C3细胞,以提供然后暴露于前体药物的MCL。
筛选试验的结果提供在图18、18.1、19、20、20.1、21和23中并且显示本发明的化合物好像抗AKR1C3的代谢,如通过在低细胞密度细胞毒性测试中在被工程化以过表达AKR1C3的HCT116和H1299细胞当与亲本野生型细胞系的测试化合物的细胞毒性比较时(分别图18和18.1)不能提供增加细胞毒性所指示。如发明人已经测定‘假阴性’可发生在该试验中,原因是代谢物扩散进入基本上无限稀释的试验培养基中,通过比较作为多细胞层(MCLs)和暴露于测试化合物生长的野生型HCT116细胞和过表达AKR1C3的HCT116细胞的克隆细胞杀伤,对于所有本发明的化合物确认了缺少AKR1C3介导的细胞毒性。这里,所有的测试化合物显示与野生型同基因的细胞系相比,在过表达AKR1C3的MCL中不产生另外的细胞杀伤,指示它们不被AKR1C3代谢成为细胞毒素代谢分子(图19)。
当在被工程化以过表达来自大肠埃希杆菌的示例性硝基还原酶基因nfsA的HCT116和H1299细胞中评估时,与亲本野生型细胞系相比(分别图20和20.1),本发明的化合物显示在低细胞密度细胞毒性试验中提供卓越的细菌硝基还原酶介导细胞杀伤。这里在表达大肠杆菌nfsA的细胞中观察到另外2至3logs的细胞杀伤。为了确认测试化合物提供旁观者细胞杀伤,在采用包含97%野生型HCT116细胞(靶细胞)和3%过表达大肠杆菌nfsA的HCT116细胞(活化剂细胞)的混合HCT116 MCL的三维高细胞密度试验中评估它们。所有的化合物表明被过表达大肠杆菌nfsA的3%HCT116细胞代谢,以产生能够扩散杀伤邻近野生型HCT116细胞的细胞毒素代谢分子的能力(图21)。
在好氧和缺氧条件下的低细胞密度细胞毒性试验中测试了本发明的化合物它们产生低氧依赖性细胞毒性的能力。使用野生型HCT116、H460、H1299和SiH细胞,以及被工程化以过表达细胞色素P450还原酶(HCT116 POR),人单电子硝基还原酶的HCT116细胞。化合物14、22、18和301显示在低氧下的细胞中提供增加细胞毒性。对于化合物14和22,当HCT116细胞过表达细胞色素P450还原酶时该作用的程度增加,指示通过该人硝基还原酶增加的低氧选择性前体药物代谢提供增加的细胞毒性(图23)。
实施例3-使用硝基还原酶文库筛选前体药物化合物
筛选来自代表12个不同酶家族的20个细菌物种的55个硝基还原酶候选物的系统发生学上多样性文库它们共同代谢靶前体药物的能力。
方法采用过在SOS报告子菌株中表达来自质粒pUCX的候选物硝基还原酶基因,如Prosser等首先描述,2010,Biochem Pharmacol 79,678-687。为了增强SOS报告子系统的灵敏度,sfiA::GFP报告子构建体整合至CDF-基质粒(其包含与pUCX兼容的复制起点),以产生pANODuet报告子质粒,用于GFP筛选。另外,删除了nfsA、nfsB、azoR和nemA基因以使背景代谢最小化,并且删除了tolC基因,以使测试化合物的流量最小化;该菌株命名为SOS-R3。通过删除mdaB、ycaK和yieF基因,以使背景代谢最小化,和通过引入转录终止子至pANODUET报告子质粒获得进一步改善。该最终的报告子菌株命名为SOS-R4。
实施例4-前体药物效力的体内评估
动物饲养
由Vernon Jansen Unit(shared vivarium,University ofAuckland)饲养专用无病原体雌性纯合子裸NIH-III(NIH-Lystbg Foxn1nu Btkxid)小鼠。动物居住在Techniplast微型隔离笼和提供标十二小时白天-黑夜光方案。动物接受标准啮齿动物膳食(Harlan Teklad膳食2018i)和水自由采食。所有动物研究由University ofAuckland Animal Ethics Committee批准。
肿瘤细胞接种
动物在肿瘤接种时重18-25g。通过接种在组织培养物中生长的细胞(1x107细胞100uL无血清α-MEM),肿瘤在小鼠的右侧皮下生长。使用电子卡尺每周监测肿瘤尺寸三次并且当肿瘤直径≥7mm时启动治疗。
生长延迟
将具有肿瘤的小鼠随机分入适当的治疗组并且记录肿瘤尺寸和体重。在实验当天配制测试化合物并且保持在箔包装的管中,避免直接荧光。如果动物的募集持续多天,药物原液等样试分至管中并且一次冷冻在-4℃中直到使用。通过腹膜内注射小鼠用单个(或BID)剂量的前体药物治疗并且其后每隔两天监测肿瘤尺寸和体重。肿瘤体积计算为π(1x w x w)/6,其中1是主轴和w是垂直附轴。当它们达到适当的生存终点或当体重减轻超过治疗前的值20%时,剔除动物。
照射和无照射的切除试验
通过接种在组织培养基中生长的细胞,肿瘤在NIH-III小鼠的侧翼皮下生长。使用电子测径器监测肿瘤。当肿瘤达到治疗尺寸时,小鼠随机化成治疗组(每组五至七只)。以单独单个(或BID)腹膜内剂量或全身照射(60Co source)5min之后施用化合物。治疗十八个小时之后,切除肿瘤,称重,切碎,酶分解,并且铺平板,以测定克隆性。计算相对于对照的克隆原/克的组织并且测试治疗效果的显著性(ANOVA与Dunnett’s)。
雌性NIH-III小鼠中,本发明的前体药物10、11、22、23、26和60在生长包含15%大肠杆菌NfsA表达细胞和85%野生型细胞的混合HCT116和H1299肿瘤异种移植物的体内效力测试的结果显示在图22、22.1、22.2、22.3、22.4和22.5中。在单个(或BID)腹腔内剂量之后,所有测试的化合物提供显著的肿瘤生长延迟,指示代谢物扩散之后的大肠杆菌NfsA的前体药物代谢提供体内旁观者细胞杀伤。
前体药物11、22、23和300在HCT116 POR和野生型SiHa肿瘤异种移植物中的体内效力测试的结果显示在图24、25和26中。这里,作为单个试剂或在单个(或BID)腹腔内剂量之后结合10或15Gy照射评估测试化合物。所有研究的前体药物表明高于单独用照射可实现的增加的细胞杀伤,指示化合物杀伤肿瘤异种移植物中缺氧(和所以抵抗照射)细胞的能力。
材料和方法
硝基还原酶基因文库
55个成员的候选物硝基还原酶文库中的全部列举如下(按细菌菌株的字母表顺序(下划线)每个扩增自):凝结芽孢杆菌(36D1)nfsA;枯草芽孢杆菌(ATCC 6051)nfrA、ycnD、ydgI、yfkO、ywrO;苏云金杆芽孢杆菌(serovar konkukian,菌株97-27)nfsA;克氏柠檬酸杆菌(ATCC 27156)nfsA、nfsB;坂崎肠杆菌(Chronobacter)(ATCC 29544)nfsA、nfsB;软腐欧文氏菌(亚种Atrosepticum SCRI1043)nfsA;大肠埃希杆菌(W3110)azoR、kefF、mdaB、nemA、nfsA、nfsB、wrbA、ycdI、ydjA、yieF;肺炎克雷伯氏菌(ATCC 13883)nemA、nfsA、nfsB、ycdI、ydjA;乳酸菌sakei(亚种sakei 23K)nfsA;威氏李斯特氏菌(serovar 6b,菌株SLCC5334)nfsA;无害李斯特菌(Clip 11262)nfsA、ywrO;耻垢分枝杆菌(菌株MC2155)nfsA;点型念珠蓝细菌(PCC 73102)nfsA;铜绿假单胞菌(PAO1)nfsB(PA5190)、nqo1(PA4975)、yieF(PA1204);恶臭假单胞菌(KT2440)azoR(PP4538)、nfsA(PP2490)、nfsB(PP2432)、nqo1(PP3720);豌豆细菌性枯萎病菌(1448a)mdaB,wrbA;伤寒沙门氏菌(ATCC 19430)azoR、nemA、nfsA、nfsB;费氏弧菌(ATCC 7744)FRaseI(黄素还原酶1),nfsA、ywrO;哈维氏弧菌(ATCC 33843)frp(黄素还原酶P)、nfsB;哈维氏弧菌(HY01)CO-frp(大肠杆菌密码子优化的黄素还原酶P变体);创伤弧菌(ATCC 27562)azoR、nfsA、nfsB、nemA。为了用相同家族名区分基因或酶,对于该工作的目的,使用标准命名提及每个氧化还原酶,随后下划线和属和种的两字母缩写,例如分别来自肺炎克雷伯氏菌的NfsA酶的NfsA_Kp和NemA_Ec和来自大肠杆菌的NemA酶。
除了筛选55个成员的候选物硝基还原酶文库,也表明了化合物14、18、22、23、24、25、26、27和64对一系列先前已经被工程化用于改善PR-104A活性的大肠杆菌NfsA(NfsA_Ec)的单突变和多突变的变体,和枯草芽孢杆菌NfrA(NfrA_Bs/NfsA_Bs)单突变的变体的的活性。NfsA_Ec的单突变的变体是NfsA_Ec_12S(12位的天然精氨酸被丝氨酸替换);NfsA_Ec_41Y(41位的天然丝氨酸被酪氨酸替换);NfsA_Ec_134A(134位的天然天冬酰胺被丙氨酸替换);和NfsA_Ec_222E(222位的天然赖氨酸被谷氨酸替换)。NfsA_Bs单突变的变体是NfsA_Bs_234P(234位的天然精氨酸被脯氨酸替换)。大肠杆菌NfsA多突变的变体是多17(I5T、S41Y、R225P、F227S)、多22(S41Y、E99G、L103M、R225P、F227S)和多42(S41Y、L103M、R225G、F227S)。
GFP-SOS试验
SOS-R4中融化55个成员的硝基还原酶文库储存的丙三醇并且用于接种过夜培养基(用0100μg ml-1氨苄西林、50μg ml-1大观霉素、0.4%葡萄糖改性的Lysogeny发酵液(LB)),其30℃,200rpm下孵育16h。第二天早上,用96孔板的单个孔中的15μl过夜培养基,通过接种195μl新鲜试验培养基(LB+100μg ml-1氨苄西林、50μg ml-1大观霉素、0.2%葡萄糖、50μM IPTG)开始GFP-SOS试验。平板在30℃下,200rpm孵育2.5h(激发前时间),其后通过迅速50∶50掺入试验培养基(+DMSO至0.5%终浓度)1∶2稀释培养物并且新鲜激发培养基(试验培养基+期望浓度的药物,DMSO至0.5%终浓度),在384孔板上一式两份至各自的80μl终体积。平板在30℃,200rpm下孵育6h(激发时间)。以激发488nm/发射509nm测定GFP表达。通过OD600测定浑浊度并且丢弃来自任何重复的不在中值浑浊度15%之内的荧光数据。
纯化的酶动力学
对于化合物14、18、22、23、24、25、26、27和64,用纯化的NfsA_Ec在400nm分光光度评估稳态酶动力学,以直接监测化合物减少,如Prosser等(2013)为PR-104A描述。测量4800M-1cm-1(化合物14和18),5200M-1cm-1(化合物22),5500M-1cm-1(化合物25和26),5600M-1cm-1(化合物23,24和27)和10,000M-1cm-1(化合物64)的摩尔消光系数(如Prosser等,2013,Biochem Pharmacol 85,1091-1103为PR-104A描述)并且用于计算酶活性。反应在60μl的UVettes(Eppendorf)中,使用2mm光路长度进行。反应包含10mM Tris-Cl(pH 7.0)、4%DMSO,0.25mM NADPH和不同的化合物浓度。通过添加6μl的酶启动反应并且测量吸光度的改变20s(在线性期间)。为了计算Km和kcat,底物浓度从~0.2x Km至多达5x Km或通过化合物溶解度预测的最大浓度变化。使用Sigmaplot 10.0(Systat software Inc.)进行非线性回归分析和Michaelis-Menten曲线拟合。
NfsB速度试验
化合物的溶解度限制意思是表观的Km和kcat参数可能未对NfsB_Ec精确测定,其对于所有的化合物在可实现的浓度范围内展示线性减少速度。但是,对于每个化合物以一个固定浓度(600μM)进行评估通过NfsB_Ec的化合物减少的相对速度。以与描述测定NfsA_Ec的纯化的酶动力学相同的方式建立各个反应。
结果
前体药物在细菌中NTR筛选和NfsA动力学和代谢试验的NfsB速度的结果提供在图17.1至17.11中。化合物14、18、22、23、24、25、26、27和64都确认为是来自数个细菌物种的NfsA和NfsB家族的多个野生型和突变体细菌硝基还原酶的底物,如通过它们被细菌硝基还原酶代谢成导致细菌中SOS应答的基因毒性代谢分子能力评估(图17.1至17.9)。通过在存在重组大肠杆菌NfsA和NADPH辅助因子的情况下孵育不同浓度的化合物并且然后跟踪辅助因子随着时间的推移的损失,确认测试化合物被大肠杆菌NfsA的代谢。酶Km’s确认为范围是160至820uM。酶kcat范围是3.6至21.9s-1(图17.10)。通过在存在重组大肠杆菌NfsB和NADPH辅助因子的情况下孵育每个化合物的一个固定浓度(600μM)并且然后跟踪辅助因子随着时间的推移的损失确认测试化合物被大肠杆菌NfsB的代谢。所有测试的化合物被大肠杆菌NfsB代谢,速度从200至4500umol/min/mg变化(图17.11)。
遍及本说明书,除非上下文另外规定,词语“包括”,“包含”等以包含性含义解释,而不是以排他性含义解释,也就是说,以“包括但不限于”的含义解释。
在本说明书中提及任何现有技术不解释为而且也不应解释为承认或任何形式的暗示现有技术形成公知常识的一部分。
应认识到本发明的化合物可以不同的几何和对映体形式存在,并且包括这些化合物的纯净形式和混合物二者。
如果有的话,上面和下面引用的所有申请、专利和出版物的内容,通过参考并入本文。
本发明可大体上认为由在说明书中单独或共同提及或指示的部分、要素和特征,两个或更多个所述部分、要素或特征的任何或所有组合而组成。
其中前述说明书已经提及具有其已知等价物的整数或组分,那些整数如同单独提出并入本文。
应当注意,本文所述的目前优选的实施方式的各种改变和修饰对本领域技术人员是显而易见的。可作出这些改变和修饰而不背离本发明的精神和范围,并且不减少其伴随的优势。所以,期望这些改变和修饰包括在本发明的范围内。
- 还没有人留言评论。精彩留言会获得点赞!