一种注射用泮托拉唑钠的制备方法与流程
本发明属于医药
技术领域:
;涉及一种泮托拉唑钠的制备方法,尤其是一种注射用泮托拉唑钠的制备方法。
背景技术:
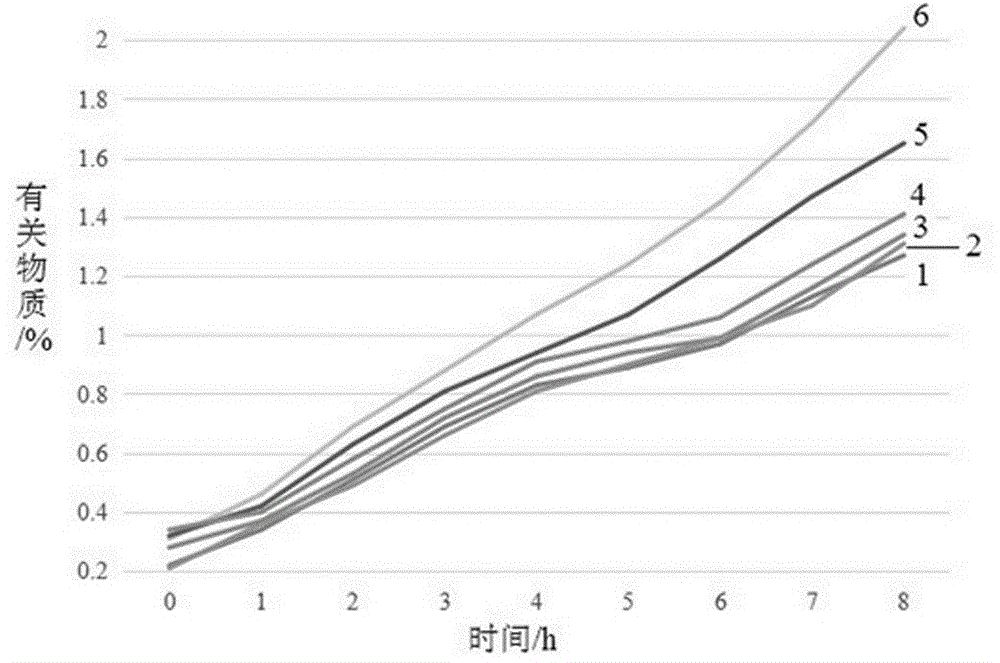
:泮托拉唑钠是第二个上市的质子泵抑制剂(protonpumpinhibitors,ppi)。泮托拉唑钠的化学名称为:5-二氟甲氧基-2-[(3,4-二甲氧基-2-吡啶基)甲基]亚磺酰基]-1h-苯并咪唑钠一水合物,其分子量为423.4。泮托拉唑钠是白色或类白色的结晶性粉末,易溶于水或甲醇,难溶于三氯甲烷或乙醚。注射用泮托拉唑钠及泮托拉唑钠原料均已收载于中国药典2010年版和2015年版第二部中。泮托拉唑钠可以选择性地抑制胃壁细胞的h+/k+-atp酶,使胃壁细胞的h+不能转运到胃中,从而强力抑制胃酸分泌。临床上广泛应用于胃溃疡、十二指肠溃疡、急性胃粘膜病变、复合性胃溃疡等急性上消化道出血。与抗生素联合用药可以根据幽门螺杆菌感染,预防和治疗非甾体抗炎药引发的病变,治疗卓-艾综合症,以及非食管静脉曲张引发的上消化道出血或非溃疡消化不良等。这是目前临床上治疗此类疾病最成功最有效的方法。然而,泮托拉唑钠化学性质不稳定,尤其对ph条件最为敏感,在ph<8.0条件下放置3-4h颜色就由无色变为微黄,随后生成淡黄色的不溶性颗粒。这些颗粒的粒径通常大于2μm,进入人体血液后,可能引起血管栓塞、过敏反应或热原样反应等不良反应。此外,泮托拉唑钠在酸性条件、受热和见光条件下容易降解,生成有关物质。中国专利申请cn101249073a公开了一种泮托拉唑钠脂质体冻干制剂,该冻干制剂由含有抗氧剂的大豆卵磷脂和胆固醇形成的脂质体包封泮托拉唑钠形成的冻干制剂。该冻干制剂可以用于静脉给药。然而,该冻干制剂的制备工艺繁琐复杂,质量控制较难,难以大规模工业化生产。另一方面,该冻干制剂酸度分布在7.3-7.8之间,但有关物质却始终保持在0.3%左右,重复试验表明,该冻干制剂的质量稳定性不佳。中国专利申请cn101548957a公开了一种泮托拉唑钠亚微乳冻干制剂。该冻干制剂由泮托拉唑钠、生物降解聚合物、乳化剂、骨架支持剂和稳定剂制成。该冻干制剂的平均粒径为50-200nm,符合静脉注射制剂的要求。该冻干制剂通过采用特定的赋形剂如生物降解聚合物、乳化剂、骨架支持剂、稳定剂等与活性成分的组合,制成亚微乳冻干制剂。然而,所使用的的聚乳酸及其共聚物脂溶性较高,导致泮托拉唑钠的包封率很低。此外,这类聚合物容易降解成乳酸和羟基乙酸等酸性产物并累计在内部,从而导致泮托拉唑钠在酸性条件下降解。另一方面,现有的冻干制剂多数需要在临用前加入专用溶媒溶解并用0.9%的nacl注射液稀释,但该溶液在注射条件下稳定性较差,一般要求在3h内注射完毕,大大限制了泮托拉唑钠在临床连续输液时的应用。因此,针对现有技术的上述缺陷,需要进一步寻找一种注射用泮托拉唑钠的制备方法。技术实现要素:本发明的发明目的是克服现有技术的不足,提供一种包封率和稳定性均较高的注射用泮托拉唑钠的制备方法。为实现上述目的,本发明提供了一种注射用泮托拉唑钠的制备方法,所述方法包括:(1)将泮托拉唑钠溶于0.5-1.5wt%的乳化剂溶液中,用碱调节ph值,得到水性溶液;(2)将改性聚乳酸羟基乙酸共聚物溶于二氯甲烷中,得到油性溶液;(3)将所述油性溶液和所述水性溶液在12000-18000rpm转速下乳化0.5-5min,得到油包水乳液;(4)将所述油包水乳液缓慢加入0.1-0.6wt%的乳化剂溶液中,6000-14000rpm下乳化5-60s,得到水包油包水乳液;(5)将所述水包油包水乳液倒入0.1-0.6wt%的乳化剂溶液中,室温下400-800rpm搅拌1-8h,除去二氯甲烷溶剂,得到悬浮液;(6)将所述悬浮液离心,用水洗涤,冷冻干燥,得到注射用泮托拉唑钠冻干制剂。根据本发明所述的制备方法,其中,所述乳化剂溶液进一步加入碱性氨基酸。优选地,所述碱性氨基酸选自精氨酸、赖氨酸和组氨酸;更优选地,所述碱性氨基酸选自精氨酸和赖氨酸;以及,最优选地,所述碱性氨基酸选自精氨酸。在一个具体的实施方式中,所述碱性氨基酸选自l-精氨酸。根据本发明所述的制备方法,其中,所述ph值为8.0-12.0。优选地,所述ph值为8.5-11.5;更优选地,所述ph值为9.0-11.0;以及,最优选地,所述ph值为9.5-10.5。在一个具体的实施方式中,所述ph值为10.0。根据本发明所述的制备方法,其中,所述改性聚乳酸羟基乙酸共聚物选自pva接枝的聚乳酸羟基乙酸共聚物。根据本发明所述的制备方法,其中,所述pva选自聚合度200-400醇解度85-92%的医用级pva。优选地,所述pva选自聚合度220-380醇解度86-91%的医用级pva;更优选地,所述pva选自聚合度250-360醇解度86-90%的医用级pva;以及,最优选地,所述pva选自聚合度280-320醇解度87-89%的医用级pva。在一个具体的实施方式中,所述pva选自聚合度300醇解度88%的医用级pva。根据本发明所述的制备方法,其中,所述改性聚乳酸羟基乙酸共聚物由pva、外消旋丙交酯和乙交酯混合物熔融聚合而成。根据本发明所述的制备方法,其中,外消旋丙交酯和乙交酯的摩尔比为1:1。根据本发明所述的制备方法,其中,所述pva与所述外消旋丙交酯和乙交酯混合物的重量比为1:(0.7-4.5)。优选地,所述pva与所述外消旋丙交酯和乙交酯混合物的重量比为1:(0.8-4);更优选地,所述pva与所述外消旋丙交酯和乙交酯混合物的重量比为1:(0.9-3.5);以及,最优选地,所述pva与所述外消旋丙交酯和乙交酯混合物的重量比为1:(1-3)。在一个具体的实施方式中,所述pva与所述外消旋丙交酯和乙交酯混合物的重量比为1:2。根据本发明所述的制备方法,其中,所述改性聚乳酸羟基乙酸共聚物的分子量mn为10000-50000,pdi为1.35-1.75。优选地,所述改性聚乳酸羟基乙酸共聚物的分子量mn为12000-48000,pdi为1.40-1.70;更优选地,所述改性聚乳酸羟基乙酸共聚物的分子量mn为14000-45000,pdi为1.45-1.65;以及,最优选地,所述改性聚乳酸羟基乙酸共聚物的分子量mn为15000-43000,pdi为1.49-1.62。在一个具体的实施方式中,所述改性聚乳酸羟基乙酸共聚物的分子量mn为27460,pdi为1.49。根据本发明所述的制备方法,其中,所述乳化剂选自聚合度200-400醇解度85-92%的医用级pva。优选地,所述乳化剂选自聚合度220-380醇解度86-91%的医用级pva;更优选地,所述乳化剂选自聚合度250-360醇解度86-90%的医用级pva;以及,最优选地,所述乳化剂选自聚合度280-320醇解度87-89%的医用级pva。在一个具体的实施方式中,所述乳化剂选自聚合度300醇解度88%的医用级pva。根据本发明所述的制备方法,其中,泮托拉唑钠、碱性氨基酸和改性聚乳酸羟基乙酸共聚物的重量比为1:(0.2-3.2):(1-3)。优选地,泮托拉唑钠、碱性氨基酸和改性聚乳酸羟基乙酸共聚物的重量比为1:(0.3-2.8):(1.2-2.8);更优选地,泮托拉唑钠、碱性氨基酸和改性聚乳酸羟基乙酸共聚物的重量比为1:(0.4-2.4):(1.4-2.6);以及,最优选地,泮托拉唑钠、碱性氨基酸和改性聚乳酸羟基乙酸共聚物的重量比为1:(0.5-2):(1.6-2.4)。在一个具体的实施方式中,泮托拉唑钠、碱性氨基酸和改性聚乳酸羟基乙酸共聚物的重量比为1:1:2。发明人发现,本发明的注射用泮托拉唑钠冻干制剂所含的改性聚乳酸羟基乙酸共聚物和精氨酸对注射稳定性增加起了关键作用。不希望局限于任何理论,经改性后,聚乳酸羟基乙酸共聚物变为双亲性聚合物,水溶性增加,导致内水相对泮托拉唑钠的亲和性显著增加,从而使得包封率提高。此外,加入的精氨酸使得注射用泮托拉唑钠冻干制剂内部的ph条件偏向碱性,从而改善了泮托拉唑钠的稳定性。与现有技术相比,本发明具有下列有益技术效果:i)本发明的注射用泮托拉唑钠在注射条件下稳定性较好,大大改善了泮托拉唑钠在临床连续输液时的应用。ii)本发明的注射用泮托拉唑钠的包封率较高。iii)本发明制备方法简单易行,重复性好,同时无需其它辅料;设备成本低且无污染;可产生巨大的社会效益和经济效益,适合普遍推广使用。附图说明图1是实施例1-3和比较例1-3注射用泮托拉唑钠冻干制剂的有关物质岁时间变化曲线(其中,曲线1-6依次为实施例1、实施例3、实施例2、比较例3、比较例2和比较例1)。具体实施方式下面结合具体实施方式,进一步阐述本发明。应理解,这些实施例仅用于说明本发明而不用于限制本发明的范围。此外应理解,在阅读了本发明的内容之后,本领域技术人员可以对本发明作各种改动或修改,这些等价形式同样落于本申请所附权利要求书所限定的范围。通过下述实施例将有助于理解本发明,但不能限制本发明的范围。在各实施例中,所使用的水为纯化水,未注明纯度的原料纯度均为色谱纯。制备实施例1:将2.6g聚合度300醇解度88%的医用级pva预先在40°c真空干燥至恒重,置于反应容器中,抽真空,通氩气,交替进行三次;然后升温至80°c,加入摩尔比例为1:1的外消旋丙交酯(2.88g)和乙交酯(2.32g)混合物,在氩气保护下加热至140°c,使反应混合物在不断搅拌下完全熔融,然后再加入0.0086g的sn(oct)2,反应6h;反应完毕后,将粗产物使用二氯甲烷作为溶剂,并使用乙醚作为反溶剂,利用溶剂交换法进行纯化,重复三次得到产物。产物即为pva接枝的聚乳酸羟基乙酸共聚物。产物冷冻干燥备用。使用凝胶渗透色谱仪和聚苯乙烯标准样品测试产物的分子量及分子量分布。结果表明,pva接枝的聚乳酸羟基乙酸共聚物的数均分子量为27640,分子量分布指数pdi=1.49。制备实施例2:将2.6g聚合度300醇解度88%的医用级pva预先在40°c真空干燥至恒重,置于反应容器中,抽真空,通氩气,交替进行三次;然后升温至80°c,加入摩尔比例为1:1的外消旋丙交酯(1.44g)和乙交酯(1.16g)混合物,在氩气保护下加热至140°c,使反应混合物在不断搅拌下完全熔融,然后再加入0.0045g的sn(oct)2,反应5h;反应完毕后,将粗产物使用二氯甲烷作为溶剂,并使用乙醚作为反溶剂,利用溶剂交换法进行纯化,重复三次得到产物。产物即为pva接枝的聚乳酸羟基乙酸共聚物。产物冷冻干燥备用。使用凝胶渗透色谱仪和聚苯乙烯标准样品测试产物的分子量及分子量分布。结果表明,pva接枝的聚乳酸羟基乙酸共聚物的数均分子量为14233,分子量分布指数pdi=1.62。制备实施例3:将2.6g聚合度300醇解度88%的医用级pva预先在40°c真空干燥至恒重,置于反应容器中,抽真空,通氩气,交替进行三次;然后升温至80°c,加入摩尔比例为1:1的外消旋丙交酯(4.32g)和乙交酯(3.48g)混合物,在氩气保护下加热至140°c,使反应混合物在不断搅拌下完全熔融,然后再加入0.013g的sn(oct)2,反应8h;反应完毕后,将粗产物使用二氯甲烷作为溶剂,并使用乙醚作为反溶剂,利用溶剂交换法进行纯化,重复三次得到产物。产物即为pva接枝的聚乳酸羟基乙酸共聚物。产物冷冻干燥备用。使用凝胶渗透色谱仪和聚苯乙烯标准样品测试产物的分子量及分子量分布。结果表明,pva接枝的聚乳酸羟基乙酸共聚物的数均分子量为43187,分子量分布指数pdi=1.56。实施例1将0.5g泮托拉唑钠和0.5gl-精氨酸溶于5ml1wt%pva溶液,并用氢氧化钠稀溶液调节至ph=10.0。其中,pva为聚合度300醇解度88%的医用级pva。然后将上述混合液加入5ml溶解有1g来自制备实施例1的pva接枝的聚乳酸羟基乙酸共聚物的二氯甲烷溶液中,在15000rpm转速下乳化1min,得到油包水乳液。然后将油包水乳液用注射器缓慢加入10ml0.3wt%pva溶液中,10000rpm下乳化15s,得到水包油包水乳液。然后将水包油包水乳液倒入80ml0.3wt%pva溶液中,室温600rpm下搅拌4h,除去二氯甲烷溶剂,得到悬浮液。离心,用水洗涤,然后冷冻干燥,得到注射用泮托拉唑钠冻干制剂。4°c下保存。实施例2将0.5g泮托拉唑钠和0.25gl-精氨酸溶于5ml1wt%pva溶液,并用氢氧化钠稀溶液调节至ph=10.5。其中,pva为聚合度300醇解度88%的医用级pva。然后将上述混合液加入5ml溶解有0.8g来自制备实施例2的pva接枝的聚乳酸羟基乙酸共聚物的二氯甲烷溶液中,在15000rpm转速下乳化1min,得到油包水乳液。然后将油包水乳液用注射器缓慢加入8ml0.3wt%pva溶液中,10000rpm下乳化10s,得到水包油包水乳液。然后将水包油包水乳液倒入60ml0.3wt%pva溶液中,室温500rpm下搅拌3h,除去二氯甲烷溶剂,得到悬浮液。离心,用水洗涤,然后冷冻干燥,得到注射用泮托拉唑钠冻干制剂。4°c下保存。实施例3将0.5g泮托拉唑钠和1.0gl-精氨酸溶于10ml1wt%pva溶液,并用氢氧化钠稀溶液调节至ph=9.5。其中,pva为聚合度300醇解度88%的医用级pva。然后将上述混合液加入10ml溶解有1.2g来自制备实施例3的pva接枝的聚乳酸羟基乙酸共聚物的二氯甲烷溶液中,在15000rpm转速下乳化1min,得到油包水乳液。然后将油包水乳液用注射器缓慢加入12ml0.3wt%pva溶液中,10000rpm下乳化10s,得到水包油包水乳液。然后将水包油包水乳液倒入120ml0.3wt%pva溶液中,室温800rpm下搅拌5h,除去二氯甲烷溶剂,得到悬浮液。离心,用水洗涤,然后冷冻干燥,得到注射用泮托拉唑钠冻干制剂。4°c下保存。比较例1将制备实施例1的pva接枝的聚乳酸羟基乙酸共聚物替换为数均分子量mn=28000的摩尔比为50:50聚乳酸羟基乙酸共聚物;其余条件同实施例1。比较例2不加入精氨酸;其余条件同实施例1。比较例3加入0.05gl-精氨酸;其余条件同实施例1。取少量实施例1-3和比较例1-3的注射用泮托拉唑钠冻干制剂加入水中,震荡使其均匀分散,使用激光粒度仪测试其粒径分布。在包封率测试中,准确称量干燥的注射用泮托拉唑钠冻干制剂10mg,加入1ml二氯甲烷溶解,并加入4ml甲醇震荡,8000rpm离心10min取上清液,经0.45μm滤膜过滤后取20μl进样,然后计算其包封率。参见表1。表1编号粒径范围/nm包封率/%实施例1350-140071.4实施例2400-150068.5实施例3350-160063.7比较例1200-120035.1比较例2200-130061.4比较例3300-140065.2将实施例1-3和比较例1-3的注射用泮托拉唑钠冻干制剂用0.9%的nacl溶液稀释至100ml,测定室温下8h内有关物质的变化。检测方法为中国药典2015版第二部第706页。结果具体参见图1。结果表明,本申请实施例1-3的注射用泮托拉唑钠冻干制剂相对于比较例1-3在加入0.9%nacl溶液配制成注射液后,稳定性大大增加。连续输液6h后,相关物质含量仍然符合药典规定的质量标准。而比较例1-3在4h后就已经不符合相关质量标准。从比较例1和2来看,本申请注射用泮托拉唑钠冻干制剂所含的pva接枝的聚乳酸羟基乙酸共聚物和精氨酸对稳定性增加起了关键作用。不希望局限于任何理论,经pva接枝改性后,聚乳酸羟基乙酸共聚物变为双亲性聚合物,水溶性增加,导致内水相对泮托拉唑钠的亲和性显著增加,从而使得包封率提高。此外,加入的精氨酸使得注射用泮托拉唑钠冻干制剂内部的ph条件偏向碱性,从而改善了泮托拉唑钠的稳定性。以上所述仅为本发明的优选实施例而已,并不用于限制本发明,对于本领域的技术人员来说,本发明可以有各种更改和变化。凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均包含在本发明的保护范围之内。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1