一种全氟酮类环保型替代气体热力学仿真方法与流程
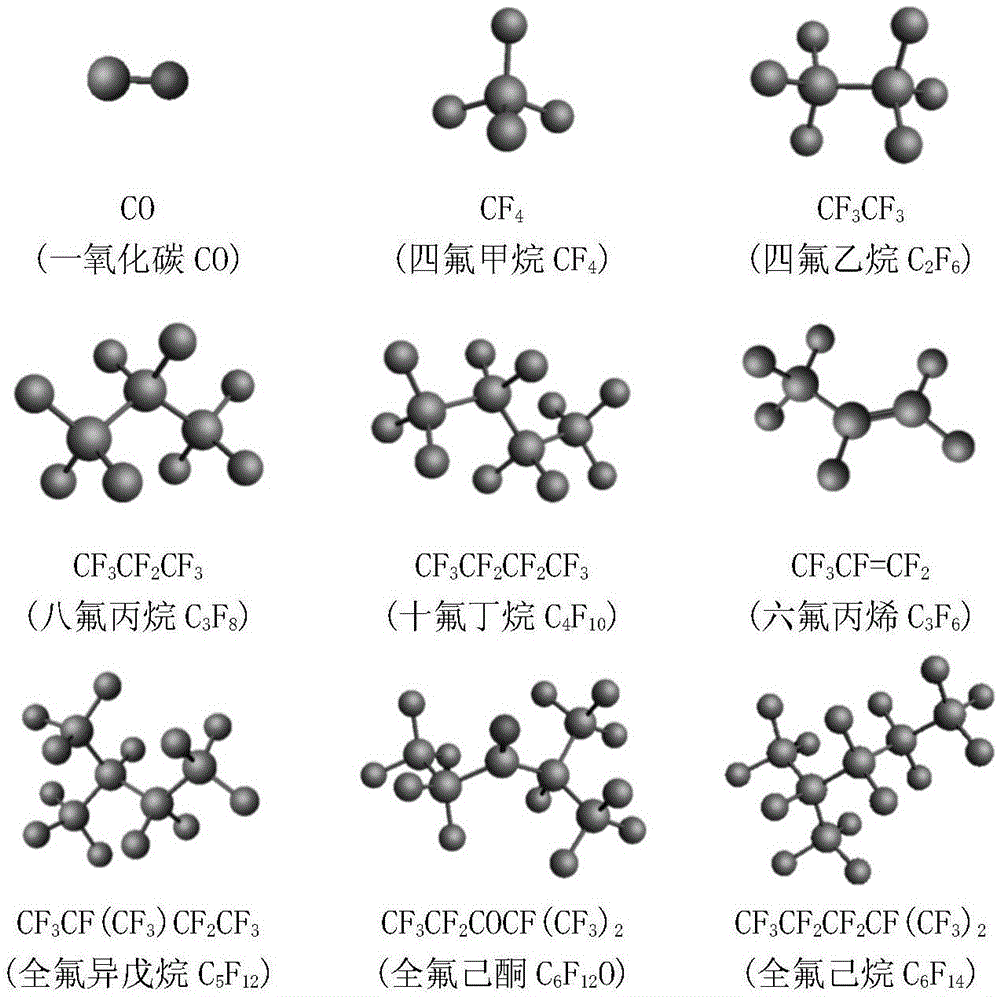
本发明属于气体绝缘电气设备内气体绝缘介质过热故障机理分析的领域,具体涉及一种全氟酮类环保型替代气体热力学仿真方法。
背景技术:
六氟化硫(sf6)因为其突出的绝缘性能和灭弧特性,目前广泛地运用于金属封闭开关设备(gasinsulatedswitchgear,gis)中,其中高压开关设备的用气量约占sf6用气量的80%以上,中压开关设备的用气量约占10%。随着电力行业的不断发展,电网系统中gis设备的安装量越来越大,sf6的使用量也迅速增加,而sf6气体绝缘设备的调试、检修的过程以及气体的泄露和回收都会使sf6进入大气环境中。根据ipcc提出的诸多温室气体的全球变暖潜能值(globalwarmingpotential,gwp),sf6的gwp值最大,500年的gwp值为32400,且由于sf6化学性质稳定,在大气中存留时间可长达3200年。因此,联合国气候变化框架公约于1997年于日本京都制定的《京都议定书》,明确规定了sf6作为六大温室气体之一,要求作为温室气体排放大户的发达国家采取具体措施限制温室气体的排放。据不完全统计,过去五年,大气中的sf6气体含量增加了约20%,而全球每年生产的sf6近80%应用于电力工业。因此,出于长久的环保考虑,减少甚至杜绝sf6使用是电力系统发展的必然。
目前,在电力系统内对环保型替代气体的研究和探索已成为近十几年国内外学者的研究热点。abb公司提出了cnf2no类全氟酮类绝缘气体在电气设备中的应用前景,其中c5f10o(c5-perfluoroketone,c5-pfk)和c6f12o(c6-perfluoroketone,c6-pfk)绝缘介质不燃不爆无毒,其gwp值接近于1,臭氧衰减指数(ozonedepletionpotential,odp)为0,且绝缘强度为纯sf6气体的2倍和2.5倍左右,特别适用于中低压柜式气体绝缘金属封闭开关设备(cubiclegas-insulatedswitchgear,c-gis)中。虽然全氟酮类绝缘气体具有十分优异的绝缘性能和环保指标,但由于其液化温度高达26.9℃和49.2℃,无法单独使用,需要和低液化温度的缓冲气体如co2、n2等混合使得液化温度降低以防止其液化。而在c-gis内部的母线、接头等常常会因为震动、腐蚀等导致有效接触面积减小,造成接头接触不良,或导体发生过载故障时,缺陷部位的热稳定将被破坏,使气体绝缘介质温度不断上升,引起局部过热性故障(partialoverthermalfault,pof)的发生。全氟酮类环保型替代气体cnf2no作为气体绝缘介质,在c-gis内部出现pof时将可能导致全氟酮类混合气体中体积分数较低的cnf2no发生热解,其中产生的分解产物可能还会对c-gis内部金属部件的表面造成腐蚀等,对绝缘性能产生危害。然而,目前针对cnf2no的研究主要在环保指标和电气强度方面具有替代sf6气体的可能性,以及其在在c-gis内部出现pof时的分解特性和热稳定性暂不明确,而这些内容均是全氟酮类环保型绝缘气体在电力系统领域发展研究的关键问题之一。
密度泛函理论(densityfunctionaltheory,dft)是量子化学、计算化学中最流行的第一性原理,其理论本身是基于量子力学基本方程——薛定谔方程而来。由于密度泛函理论(densityfunctionaltheory)在计算量和计算精度上的优越性,因此在计算模拟领域具有实际的应用价值,应用广泛且历史悠久,可用于理论化学方面,如预测热化学数据、计算分子静态参数、优化分子结构、以及探讨过渡态结构和活化能等方面。因此,通过仿真分析的手段对其过热分解的机理进行理论性的研究工作,可以在热力学的基础上初步地掌握全氟酮类环保型绝缘气体过热分解的规律,对今后现场实验具有指导意义。
技术实现要素:
本发明基于密度泛函理论针对全氟酮类环保型绝缘气体在过热情况下分解机理的热力学研究。在跨尺度计算化学平台(amsterdammodelingsuite,ams)中的构建全氟酮类环保型绝缘气体及过热分解产物的分子结构,基于密度泛函理论中的hybrid:b3lyp方法对其结构进行优化、通过各个分子片段的能量计算cnf2no分子间键能,然后根据体系内反应物和产物的反应热来分析cnf2no热分解的反应机理。该分析方法可以对全氟酮类环保型绝缘气体在过热故障发生的情况下,气体分子发生热分解的热力学研究进而分析cnf2no热分解的规律,为进一步发展全氟酮类环保型绝缘气体在中低压柜式气体绝缘金属封闭开关设备中使用打下理论基础。
本发明的技术方案是:
一种全氟酮类环保型替代气体热力学仿真方法,其特征在于,针对全氟酮类环保型绝缘气体再过热故障情况下的分解产物进行仿真分析,具体包括:
步骤1、初步构建分子结构:在ams(amsterdammodelingsuite)平台的gui界面,根据全氟酮类环保型绝缘气体和热分解产物的分子式构建相应的分子结构,先使用半经验分子轨道程序包mopac中的非限制hartree-fock(uhf)方法,对气体分子的结构进行初步优化;
步骤2、dft泛函的选择:在优化本体系中全氟酮类环保型绝缘气体及其分解产物分子结构和计算反应过程中的反应热时使用m06-2x泛函方法,而在计算全氟酮cnf2no分子间键能的时候使用b3lyp-d3(bj)泛函方法;
步骤3、基组的选择:基组从sz到dz、dzp、tzp、tz2p到qz4p,其中d、t、q表示double、triple、quadruple,(n)p表示n个极化函数,基组逐渐增大;基组增大到一定程度之后,计算量也迅速的增大,但对结果的改变就逐渐趋近于零,称之为基组的收敛;全氟酮类环保型绝缘气体及其分解产物进行密度泛函计算时选择tzp基组;
步骤4、分子结构的优化:在dft方法和基组的选择基础上,对全氟酮类环保型绝缘气体及其分解产物分子进行结构优化时,选择m06-2x方法和tzp基组,对半经验分子轨道程序mapoc-uhf方法初步优化后的分子结构进行更精确的优化;在优化过程中,软件程序会不断地计算不同的分子结构,其中有五个收敛标准分别为能量变化(energychange)小于0.001、势能面的最大约束梯度(constrainedgradientmax)小于0.001、均方约束梯度(constrainedgradientrms)小于0.0006667、最大步长(cart.stepmax)小于0.01、均方步长(cart.steprms)小于0.006667,其中第二个标准最重要般也是最后一个达到的,对于大量过渡金属参与的化学反应,过渡态搜索,有可能达不到这样严格的标准,到0.003左右认为收敛;结构优化过程默认为30步,若满足五个收敛条件则停止计算,否则将会完成完整的30步过程;
步骤5、键能的计算:分子的能量由分子各个片段及连接各个片段之间的键能所构成,其中键能又是由片段间的轨道作用能、pauli作用能、静电作用能所构成,如式(1)所示;
式中,δetotal表示体系中分子的总能量,δedist表示分子片段的能量,δeelstat表示两个片段间的静电作用能,δepauli表示两个片段间的pauli作用能,δeorb表示两个片段间的轨道作用能,δebond表示分子的总键能;
计算全氟酮cnf2no分子内某个键的键能时,首先使用b3lyp-d3(bj)泛函方法和tzp基组计算全氟酮cnf2no分子的能量,再将全氟酮cnf2no分子内某个键断裂分成两个片段,然后分别对两个片段进行能量计算,最终将全氟酮cnf2no分子在体系中的能量减去该键两端分子片段在体系中的能量就是该键的能量;
步骤6、反应热的计算:化学反应中,物质发生化学变化的同时,还伴随有能量的变化,通常以热能的形式表现出来,称为反应热;如果一个化学反应中,反应物的总能量大于产物的总能量,则该反应就是放热反应,此时的δh<0,反之则为吸热反应,δh>0;
在ams平台中计算反应热有两种方式,第一种是利用键能计算反应热,第二种是利用反应前后分子的能量差进行计算;利用键能计算反应热,是把拆开分子内某化学键所吸收的能量看成该化学键的键能,计算方法为δh=ebond(反应物)-ebond(生成物),即反应热等于反应物的键能总和与生成物键能总和之差;由反应物和生成物的总能量计算反应热,计算方法为δh=生成物总能量-反应物的总能量;
针对反应物和生成物均为分子形态的化学反应,采用第二种方法对其反应热进行计算,选择m06-2x泛函方法和tzp基组,并设置反应体系的温度值t,分别对化学反应前后的反应物和生成物进行分子结构能量计算,得到其在相应体系内的能量,然后计算得到该反应途径的反应热;针对反应物和生成物存在自由基形态的化学反应,
采用第一种方法对其反应热进行计算,选择b3lyp-d3(bj)泛函方法和tzp基组,并设置反应体系的温度值t和自由基的不成对电子数,计算反应前后体系的总键能差值,该差值即为该反应途径的反应热;最后以反应热作为判据,判断该反应途径吸热或放热的程度来判断其是否会自发进行。
在上述的一种全氟酮类环保型替代气体热力学仿真方法,c5f10o热分解的产物为cf4、c2f6、c3f8、c4f10、c3f6、c6f14、co;c6f12o热分解的主要产物为cf4、c2f4、c2f6、c3f6、c3f8、c4f8、c4f10、c5f12、c6f14、co。
在上述的一种全氟酮类环保型替代气体热力学仿真方法,步骤4中,将优化过程固定为30步,若最终计算收敛,则在完整的优化过程中选取能量变化和最大约束梯度都较优的结构,否则重新进行结构优化;在完成结构优化后,通常会进行频率计算,检查是不是存在虚频,若存在虚频,则表示优化不精确,需要继续优化;完成上述优化过程后,选取最优的分子结构,导出其.xyz坐标文件作为分子最低能量的结构,以便后续键能和反应热的计算。
本发明具有如下优点:1、由于实际实验的条件无法做到绝缘气体的完全纯净,该方法能够通过反应热力学对全氟酮类环保型绝缘气体在过热时发生分解的途径进行机理分析,进而得出全氟酮类cnf2no气体分子在不同温度下时过热分解的产物。2、能够从微观层面分析全氟酮类cnf2no气体分子及其分解产物在受热情况下的能量情况。
附图表说明
图1为本发明方法优化后的反应物和分解产物分子结构。
图2为本发明方法的c6f12o分子结构示意图。
图3为本发明方法的c6f12o过热体系中反应物及分解产物分子结构能量。
图4为本发明方法的c6f12o过热体系中自由基总键能。
图5为本发明方法的c6f12o过热体系中化学反应的反应热。
图6是本发明的方法流程图。
具体实施方式
下面结合具体实施方式,进一步说明本发明。
一、首先介绍本发明的方法原理,具体包括:
1、产物确定:全氟酮类环保型绝缘气体再过热故障情况下的分解产物由课题组及国内其他课题组做过相关的实验下获取,c5f10o热分解的主要产物为cf4、c2f6、c3f8、c4f10、c3f6、c6f14、co;c6f12o热分解的主要产物为cf4、c2f4、c2f6、c3f6、c3f8、c4f8、c4f10、c5f12、c6f14、co。
2、初步构建分子结构:在ams(amsterdammodelingsuite)平台的gui界面,根据全氟酮类环保型绝缘气体和热分解产物的分子式构建相应的分子结构,先使用半经验分子轨道程序包mopac中的非限制hartree-fock(uhf)方法,对气体分子的结构进行初步优化,提供一个对称性较为理想的分子结构,以减少后续密度泛函理论方法优化的计算步数,提高优化效率。
3、dft泛函的选择:由于通常研究的分子、块状材料等电子数都高达几百甚至几万,从而薛定谔方程变成了一个无法系统地求解的多体方程。hohenberg-kohn定理给出薛定谔方程中基态能量是电子密度的函数的结论,但函数的形式却没有给出。最早的时候,人们采用了最简单的自由电子气的能量与密度的函数关系(lda),并取得了取得了巨大的成功。但因为lda泛函是采用均匀自由电子气模型,分子中电子密度变化较大,因此在非周期性体系(分子、二聚体、团簇)中很少使用。除lda之外,如今也有了gga、metagga、hybrid、metahybrid等大量泛函的出现。因此,选择一个合适的dft泛函来描述电子之间的相关交换能,是精确描述和研究反应体系的第一步。由于计算机运行效率的受限,通常情况下,计算体系分子振动、频率时使用b3lyp;计算有机分子结构优化和有机体系热力学数据(包括反应能、构型能量差、能垒等)时使用m06-2x,若加上dft-d3校正计算效果将有所改善;计算卤键时使用m06-2x;其它情况或模棱两可的时候首选b3lyp,若加上dft-d3(bj)校正效果将更好。因此,在优化本体系中全氟酮类环保型绝缘气体及其分解产物分子结构和计算反应过程中的反应热时使用m06-2x泛函方法,而在计算全氟酮cnf2no分子间键能的时候使用b3lyp-d3(bj)泛函方法。
4、基组的选择:基组在密度泛函计算中是用来展开电子的波函数。因此基组规模越大,展开波函数的自由度就越大,而密度泛函理论是一个变分理论,自由度越大也意味着在理论上越正确。基组从sz到dz、dzp、tzp、tz2p到qz4p(d、t、q表示double、triple、quadruple,(n)p表示n个极化函数),基组逐渐增大。基组增大到一定程度之后,计算量也迅速的增大,但对结果的改变就逐渐趋近于零,称之为基组的收敛。因此,在对全氟酮类环保型绝缘气体及其分解产物体系进行计算时,需要选择一个在计算量、计算的可靠程度方面都能够接受的基组。值得一提的是,计算结果的可靠程度指的是与理论极限值的接近程度,并不代表与实验值接近,理论极限值与实验值的符合情况是由所选的泛函理论方法的优劣决定。从计算的经验来看,对于比较轻的元素,例如chon之类,几何优化一般使用dzp基组足够;较重一些的元素,例如第三周期后面的元素、第四周期可以使用tzp元素是足够的;更重的元素,例如第五、六周期,使用tz2p是足够的;超重元素可以使用tz2p或者qz4p。本体系中全氟酮类环保型绝缘气体及其分解产物所所含元素c、f、o属于第二周期,因此在进行密度泛函计算时选择tzp基组就足够了。
5、分子结构的优化:在上述dft方法和基组的选择基础上,对全氟酮类环保型绝缘气体及其分解产物分子进行结构优化时,选择m06-2x方法和tzp基组,对半经验分子轨道程序mapoc-uhf方法初步优化后的分子结构进行更精确的优化。在优化过程中,软件程序会不断地计算不同的分子结构,其中有五个收敛标准分别为能量变化(energychange)小于0.001、势能面的最大约束梯度(constrainedgradientmax)小于0.001、均方约束梯度(constrainedgradientrms)小于0.0006667、最大步长(cart.stepmax)小于0.01、均方步长(cart.steprms)小于0.006667,其中第二个标准最重要,一般也是最后一个达到的,对于大量过渡金属参与的化学反应,过渡态搜索,有可能达不到这样严格的标准,一般到0.003左右也可认为收敛。结构优化过程默认为30步,若满足五个收敛条件则停止计算,否则将会完成完整的30步过程。本发明将优化过程固定为30步,若最终计算收敛,则在完整的优化过程中选取能量变化和最大约束梯度都较优的结构,否则重新进行结构优化。在完成结构优化后,通常会进行频率计算,检查是不是存在虚频,若存在虚频,则表示优化不精确,需要继续优化。完成上述优化过程后,选取最优的分子结构,导出其.xyz坐标文件作为分子最低能量的结构,以便后续键能和反应热的计算。
6、键能的计算:分子的能量由分子各个片段及连接各个片段之间的键能所构成,其中键能又是由片段间的轨道作用能、pauli作用能、静电作用能所构成,如式(1)所示。
式中,δetotal表示体系中分子的总能量,δedist表示分子片段的能量,δeelstat表示两个片段间的静电作用能,δepauli表示两个片段间的pauli作用能,δeorb表示两个片段间的轨道作用能,δebond表示分子的总键能。
因此计算全氟酮cnf2no分子内某个键的键能时,首先使用b3lyp-d3(bj)泛函方法和tzp基组计算全氟酮cnf2no分子的能量,再将全氟酮cnf2no分子内某个键断裂分成两个片段,然后分别对两个片段进行能量计算,最终将全氟酮cnf2no分子在体系中的能量减去该键两端分子片段在体系中的能量就是该键的能量。
7、反应热的计算:化学反应中,物质发生化学变化的同时,还伴随有能量的变化,通常以热能的形式表现出来,称为反应热。如果一个化学反应中,反应物的总能量大于产物的总能量,则该反应就是放热反应,此时的δh<0,反之则为吸热反应,δh>0。
在ams平台中计算反应热有两种方式,第一种是利用键能计算反应热,第二种是利用反应前后分子的能量差进行计算。利用键能计算反应热,是把拆开分子内某化学键所吸收的能量看成该化学键的键能,计算方法为δh=ebond(反应物)-ebond(生成物),即反应热等于反应物的键能总和与生成物键能总和之差;由反应物和生成物的总能量计算反应热,计算方法为δh=生成物总能量-反应物的总能量。
因此,针对反应物和生成物均为分子形态的化学反应,采用第二种方法对其反应热进行计算,选择m06-2x泛函方法和tzp基组,并设置反应体系的温度值t,分别对化学反应前后的反应物和生成物进行分子结构能量计算,得到其在相应体系内的能量,然后计算得到该反应途径的反应热;针对反应物和生成物存在自由基形态的化学反应,采用第一种方法对其反应热进行计算,选择b3lyp-d3(bj)泛函方法和tzp基组,并设置反应体系的温度值t和自由基的不成对电子数,计算反应前后体系的总键能差值,该差值即为该反应途径的反应热。最后以反应热作为判据,判断该反应途径吸热或放热的程度来判断其是否会自发进行。
二、具体案例。
下面以c6f12o环保型替代气体来验证本发明。首先根据相关文献和实验得到其过热状态下的主要分解产物为cf4、c2f4、c2f6、c3f6、c3f8、c4f8、c4f10、c5f12、c6f14、co;然后,选择m06-2x泛函方法和tzp基组对c6f12o分子及其分解产物的分子结构进行优化,并得到它们在该体系下的能量;其次,将c6f12o分子内各个键断裂,生成不同的自由基,并计算自由基的总键能;最后,综合上述结果,计算体系中各个化学反应的反应热。
本文中所描述的具体实施例仅仅是对本发明精神作举例说明。本发明所属技术领域的技术人员可以对所描述的具体实施例做各种各样的修改或补充或采用类似的方式替代,但并不会偏离本发明的精神或者超越所附权利要求书所定义的范围。
- 还没有人留言评论。精彩留言会获得点赞!