海藻酸胺化衍生物/细菌纤维素纳米晶复合凝胶构筑的功能性药物缓释医用敷料的制作方法
本发明涉及一种医用敷料,特别涉及一种海藻酸胺化衍生物/细菌纤维素纳米晶复合凝胶构筑的功能性药物缓释医用敷料,属于医疗技术领域。
背景技术:
海藻酸盐医用敷料的发展已经有30多年的历史,但是开发应用最多的还是海藻酸盐纤维敷料,并已有多家单位注册生产,如佛山优特、泰州榕兴、青岛明月等。海藻酸盐纤维敷料吸收效果好,产品性能稳定,但在使用中也存在一些不足,主要表现为不能用于结痂伤口的护理、加工流程较长、所需成本较高等。而海藻酸盐凝胶敷料产品的出现,扩大了海藻酸盐在医用敷料领域的应用范围,这类新兴敷料加工流程较短,促进伤口愈合效果显著。尽管海藻酸盐生物材料因缺乏哺乳动物细胞识别位点,具有相对生物惰性,同时具有在二价金属离子介导下温和凝胶化的特性,使其能够广泛应用于医用敷料中,但是单一的海藻酸盐凝胶由于其自身的结构和性能缺陷,它在促进伤口愈合方面能力有限,很难完全满足理想创面辅料的应用要求。海藻酸盐凝胶的这些缺陷主要体现在:(1)海藻酸盐的强亲水性影响其离子交联凝胶的结构稳定性,其凝胶结构在生理环境中容易遭受破坏;(2)海藻酸盐凝胶力学性能差,难以承受创面生理负荷;(3)海藻酸盐凝胶缺乏表面活性,易发生创面和敷料的黏着,产生二次伤害。它们严重制约了海藻酸盐凝胶敷料的应用和发展。
通过研究资料表明,海藻酸盐的钙离子交联方式是影响医用敷料结构和形貌的主要因素。由于海藻酸钙凝胶中释放出的ca2+与血液中的na+进行交换,具有促进止血的作用,因此,海藻酸盐钙离子交联的均一性对医用敷料的凝血性具有重要影响。但是海藻酸盐自身亲水性极强,所形成的海藻酸钙凝胶具有溶胀不可控的特性,在生理环境下,易发生na+-ca2+交换,使得离子交联凝胶快速崩解。因而,海藻酸盐的强亲水性是影响其离子交联水凝胶结构稳定性的主要原因之一,而且它也严重限制其实际应用。由于海藻酸盐分子链上含有大量可被修饰的羟基和羧基基团,通过合理的化学偶联方法将适量的疏水侧基接枝到其主链上,可以通过疏水侧基的疏水缔合作用自组装形成疏水微畴来改善凝胶材料的结构稳定性。特别是采用氧化-还原胺化反应制备海藻酸胺化衍生物过程中,高碘酸钠的氧化不仅使海藻酸钠的部分糖单元上的惰性羟基转变为活性醛基,显著地提高了海藻酸盐的还原胺化反应活性,而且羧基基团的保留保证了其离子交联水凝胶的形成能力。疏水侧基的接枝赋予了海藻酸盐独特的亲水亲油性能,在疏水缔合作用下可自组装形成具有疏水微腔的胶束结构,能够实现对多种药物、抗菌剂、蛋白质、基因等的包载。将药物与医用敷料相结合,通过一定的技术在医用敷料中添加药物,这在整个伤口治愈过程中发挥了举足轻重的作用:作为一种抗菌剂,可以防止细菌感染;作为一种生长剂,可以有效促进组织再生;作为一种填充剂,能够加快伤口愈合。这种将药物按照预设的量以某一速度集中于病变部位释放,既能提高药品的作用率,又能减少对正常组织器官的损伤。
而且海藻酸盐具有良好的共容性,与多种物质共混可起到协同增效的作用。学者们通常通过物理共混的方法将几种材料组合,综合考虑材料间的优缺点,互相取长补短,形成复合型材料。它们在力学性能、透水透气性、吸液速率、保湿性以及生物活性、生物相容性等方面比单一的凝胶敷料更加优异,能够满足不同创伤情况的要求,在医用敷料领域具有独特的优势。细菌纤维素纳米晶具有不同于传统材料的各种特性,包括特殊的形态和几何尺寸,高结晶度、高比表面积、流变性能、液晶行为、机械强化性能、阻隔性能、生物降解性和生物相容性等。作为一种理想的填料或补强剂,用于改善海藻酸盐凝胶通常存在的机械强度差、形态结构不佳等问题。
然而海藻酸盐相对的生物惰性致使海藻酸盐凝胶缺乏表面活性,其抗菌、促伤口愈合能力较差。为此,亟需设计一种方法,并选取抗菌活性和促伤口愈合能力优良的天然聚合物材料,以一种可控的方式修饰凝胶材料表面。
技术实现要素:
本发明利用我国优势的海藻资源,将海藻酸盐的疏水衍生化、物理共混和层层组装方法紧密结合起来,设计开发一种海藻酸胺化衍生物/细菌纤维素纳米晶复合凝胶构筑的功能性药物缓释医用敷料,同时解决单一海藻酸盐凝胶在创面敷料应用上的缺陷问题,突破技术瓶颈,为可再生资源的开发利用和医用敷料的研发提供技术支撑。
本发明解决其技术问题所采用的技术方案是:
一种海藻酸胺化衍生物/细菌纤维素纳米晶复合凝胶构筑的功能性药物缓释医用敷料的制备方法,该制备方法包括如下步骤:
(1)以高碘酸钠为氧化剂,制备理论氧化度为10%~40%的醛基海藻酸衍生物;然后以辛胺为改性剂,在氰基硼氢化钠的还原作用下制得具有两亲性的海藻酸胺化衍生物(raoa);
(2)以caco3/gdl为raoa的离子交联体系,以硫酸水解细菌纤维素制得的细菌纤维素纳米晶(bcns)作为补强剂,通过物理共混的方法将抗生素药物负载到海藻酸胺化衍生物中,在caco3/gdl的离子交联作用下构建海藻酸胺化衍生物/细菌纤维素纳米晶复合凝胶基质;
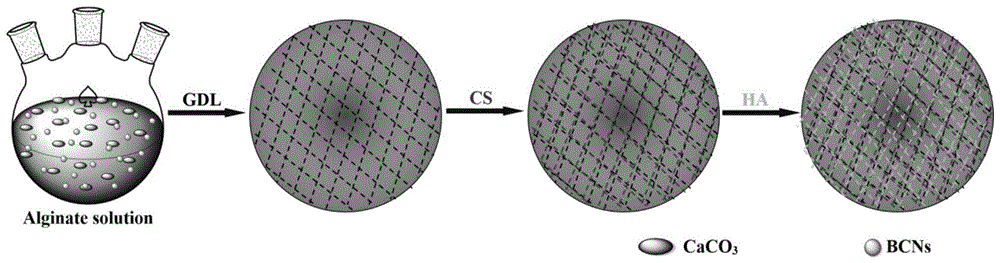
(3)结合冷冻干燥技术,通过层层组装方法将带正电的壳聚糖(cs)和带负电的透明质酸(ha)依次覆积在基质材料的表面,经过化学交联剂的交联从而制得功能性药物缓释医用敷料。上述制备流程如图1所示。
本发明的技术创新主要体现在如下几点:
(1)提出了通过氧化-还原胺化反应方法对海藻酸盐进行适当的衍生化,不仅保留了海藻酸盐的离子交联特性,而且提高了海藻酸盐离子交联水凝胶的结构稳定性,并赋予其凝胶材料良好的载药性能;
(2)利用生物大分子所带异种电荷的静电作用力,通过层层组装技术以一种可控的方式实现了材料表面的活化,有效地提高了海藻酸盐创面敷料的机械性能、抗菌性和促进伤口愈合能力;
(3)提出了以高结晶度的细菌纤维素纳米晶作为补强剂,通过物理共混方式促进凝胶材料结构稳定性和机械性能的提高。该方法为天然可再生生物大分子的开发和可持续应用提供了思路。
本发明制备的海藻酸胺化衍生物/细菌纤维素纳米晶复合凝胶功能性药物缓释医用敷料将氧化-还原胺化反应制得的两亲性海藻酸胺化衍生物的凝胶性能和载药性能结合起来,可以实现对抗生素药物的控制释放。抗生素类药物的负载和活性生物大分子ha的表面覆积,使该医用辅料表现出优异的细胞相容性和良好的抗菌性能,可实现创面的无瘢痕愈合。bcns的掺入和层层自组装过程消除了单一的海藻酸盐水凝胶在生物应用中的应力缺陷,提高了材料的机械强度。同时抗生素类药物的缓慢释放,可避免频繁换药给患者带来的痛苦。
在种类繁多的生物辅料中,作为聚阳离子电解质的壳聚糖对不同微生物如细菌、真菌和病毒表现出良好的抗菌活性。并且,壳聚糖能有效解聚释放n-乙酰基-d-氨基葡萄糖,在伤口愈合过程中促进成纤维细胞增殖,有助于更快的伤口愈合。而透明质酸作为一种天然的聚阴离子电解质,因其具有生物降解性和生物相容性并且支持细胞生长和增殖而被广泛应用于医用敷料的制备中。值得注意的是,已有研究称胎儿组织的无瘢痕修复与透明质酸的长期存在有相关性,同时透明质酸富集的环境可抑制负责瘢痕形成的基质细胞,可实现创面的无瘢痕愈合。由于海藻酸盐与壳聚糖,壳聚糖与透明质酸之间带有相反电荷,它们能够逐层交替地覆积在海藻酸盐基质材料表面,形成最外层为透明质酸的复合凝胶层。正是壳聚糖和透明质酸的先后覆积,不仅改善了海藻酸盐敷料的抗菌活性、促愈合能力,而且也提高材料的机械强度和结构完整性。这种将海藻酸盐的疏水衍生化,细菌纤维素纳米晶的补强,以及活性生物大分子间的层层自组装结合起来制备功能性药物缓释医用敷料的方法,目前还未见报道。
本发明所制备的医用敷料具有良好的3d形态和均匀的孔隙结构,能够实现对抗生素药物的控制释放,可避免频繁换药给患者带来的痛苦。而且它具有优异的细胞相容性和良好的抗菌性能,加工流程较短,促进伤口愈合效果显著,可实现工业化生产。
作为优选,步骤(2)的具体过程是:将raoa溶于水中制备raoa溶液,将细菌纤维素纳米晶(bcns)、caco3纳米粉和抗生素药物加入该raoa溶液中,混合均匀得到溶液a,然后加入葡萄糖酸内酯(gdl)引发离子交联,得到凝胶;所得凝胶置于cacl2水溶液中浸泡冲洗,以除去未参与反应的化合物和杂质,最后冷冻干燥得到海藻酸胺化衍生物/细菌纤维素纳米晶复合凝胶基质。冷冻干燥的条件一般是:先在冰箱中于-18℃下预冻30min,后转入冷阱中于-50~60℃下冷冻5~6小时。进一步的,步骤(2)中,bcns在溶液a中的质量分数为0.25%~1.0%。
步骤(2)中caco3与gdl的摩尔比为1:2,离子交联体系中ca元素与raoa中–cooh的摩尔比为0.18~0.54。
作为优选,步骤(2)中所述raoa的取代度为10%~30%,raoa溶液的质量浓度为1.5%~3.0%。
作为优选,步骤(3)的具体过程是:将干燥的海藻酸胺化衍生物/细菌纤维素纳米晶复合凝胶基质浸入壳聚糖(cs)溶液中,孵化30~60min后取出,在水中浸泡冲洗多次以除去未覆积的cs,直至洗液的ph值达到中性为止;
清洗后的凝胶经冷冻干燥后浸入透明质酸(ha)溶液中,孵化30~60min后取出,在水中浸泡并反复冲洗,最后将清洗干净的复合凝胶投入化学交联剂溶液中孵育过夜,然后取出,用超纯水反复清洗,得到的产物经冷冻干燥即得所述的功能性药物缓释医用敷料;步骤(3)中cs和ha溶液的质量浓度分别为0.5%~1.5%和1.0%~3.0%;
所述的化学交联剂选自edc/nhs混合交联剂、京尼平或戊二醛中的一种或多种。
作为优选,所述bcns是以发酵的海南椰子水作为液态培养基,通过木醋杆菌(acetobacterxylinum)合成的高结晶度的细菌纤维素,采用硫酸水解和过氧化氢氧化的方法制备的一种具有羧基官能团的纤维素纳米晶,其反应方程式如下所示:
作为优选,所述负载的抗生素药物为庆大霉素、氧氟沙星或环丙沙星中的一种或多种,其在溶液a中质量分数为0.01%~0.1%。
作为优选,所述的海藻酸胺化衍生物的制备方法如下:
(1)海藻酸钠(sa)用适量水溶解后与无水乙醇、高碘酸钠混合,避光充分搅拌,得到反应液,反应液中海藻酸钠浓度是0.5~2.5%;向反应液中加适量乙二醇避光磁力搅拌以终止反应;用氯化钠和无水乙醇沉淀终止反应后的溶液;
将所得沉淀溶解在蒸馏水中,以氯化钠和无水乙醇沉淀该溶液;如此重复沉淀3次后,将最终所得溶液装入截留分子量为3500的透析袋中透析,经冷冻干燥得到干燥的高碘酸钠氧化海藻酸衍生物;
所述氯化钠和无水乙醇的比例为1g:150~200ml;
(2)将步骤(1)制得的高碘酸钠氧化海藻酸衍生物溶解在水中,与烷基胺的甲醇溶液混合,充分反应后,加入氰基硼氢化钠,于室温下搅拌反应至充分;将所得反应液装入截留分子量为8000的透析袋中透析,经冷冻干燥得到海藻酸胺化衍生物(raoa)。
作为优选,所述海藻酸钠(sa)的质均分子量mw≥200000,其单体古洛糖醛酸(g)和甘露糖醛酸(m)的摩尔比g/m≥1.5;所述烷基胺为己胺、辛胺或癸胺中的一种。
一种所述的方法制得的海藻酸胺化衍生物/细菌纤维素纳米晶复合凝胶构筑的功能性药物缓释医用敷料。
与现有技术相比,本发明的有益效果在于:
1、本发明所述医用敷料主要基于海藻酸胺化衍生物/细菌纤维素纳米晶复合凝胶,将氧化-还原胺化反应制得的两亲性海藻酸胺化衍生物的凝胶性能和载药性能结合起来,使得负载的抗生素类药物能够缓慢的释放,可避免频繁换药给患者带来的痛苦。而且既能提高药物的作用率,又能减少对正常组织器官的损伤;
2、钙离子对raoa的均相交联,可以有效地提高该医用敷料的凝血性能;
3、由于抗生素类药物的负载和活性生物大分子ha的表面覆积,使该医用辅料表现出优异的细胞相容性和良好的抗菌性能,可实现创面的无瘢痕愈合;
4、采用物理共混方法将具有生物活性的细菌纤维素纳米晶掺入海藻酸盐凝胶基体中,消除了单一的海藻酸盐水凝胶在生物应用中的应力缺陷,提高了材料的机械强度。另外本发明加工流程较短,促进伤口愈合效果显著,可实现工业化生产。
附图说明
图1为基于海藻酸胺化衍生物/细菌纤维素纳米晶复合凝胶构筑的功能性药物缓释医用敷料的制备流程示意图;
图2为以caco3/gdl为交联体系,bcns为补强剂,通过raoa的内源交联制备的负载氧氟沙星的raoa/bcns复合凝胶功能性药物缓释医用敷料的实物图和剖切面扫描电镜图,其中(a)为未干燥raoa/bcns复合凝胶功能性药物缓释医用敷料、(b)干燥raoa/bcns复合凝胶功能性药物缓释医用敷料和(c)raoa/bcns复合凝胶功能性药物缓释医用敷料在层层组装过程中的实物图;(d)~(i)为不同bcns添加量的raoa/bcns复合凝胶功能性药物缓释医用敷料扫描电镜图:(d)0.25%、(e)0.4%、(f)0.5%、(g)0.75%、(h)0.9%和(i)1.0%;
图3为添加不同bcns条件下raoa/bcns复合凝胶功能性药物缓释医用敷料的抗压强度变化图,其中a至f为不同bcns的添加量:(a)0.25%、(b)0.4%、(c)0.5%、(d)0.75%、(e)0.9%和(f)1.0%;
图4为l929细胞在raoa/bcns复合凝胶功能性药物缓释医用敷料上分别培养2天和5天后的细胞增殖变化图,其中control为空白对照组、a至f为添加不同bcns条件下raoa/bcns复合凝胶功能性药物缓释医用敷料的细胞增殖情况:(a)0.25%、(b)0.4%、(c)0.5%、(d)0.75%、(e)0.9%和(f)1.0%。
具体实施方式
下面通过具体实施例,对本发明的技术方案作进一步的具体说明。应当理解,本发明的实施并不局限于下面的实施例,对本发明所做的任何形式上的变通和/或改变都将落入本发明保护范围。
在本发明中,若非特指,所有的份、百分比均为重量单位,所采用的设备和原料等均可从市场购得或是本领域常用的。下述实施例中的方法,如无特别说明,均为本领域的常规方法。
以下实施例中,
所述的raoa/bcns复合凝胶构筑的功能性药物缓释医用敷料,采用磷酸缓冲液(pbs)来模拟生理环境,考查其在生理环境下的凝胶稳定性;
所述的raoa/bcns复合凝胶构筑的功能性药物缓释医用敷料,以革兰氏阴性菌atccno.8739大肠杆菌和革兰氏阳性菌atccno.6538金黄色葡萄球菌作为测试菌种,考察其抗菌活性;
所述的raoa/bcns复合凝胶构筑的功能性药物缓释医用敷料,以磷酸缓冲液(pbs)作为释药介质,进行模拟释药试验,考察其对抗生素类药物的控释性能。
acetobacterxylinum细菌,来自中国普通微生物菌种保藏管理中心,保藏编码为cgmcc5173。
实施例bcns的制备
量取100ml自然发酵椰子水,向其中加入1.0g蔗糖、0.5g硫酸铵、0.05g硫酸镁和0.15g磷酸二氢钾,充分混合制成发酵培养基。在接种acetobacterxylinum细菌之前,上述发酵培养基在120℃高温灭菌30min。在ph=4.0,室温30℃条件下,静置培养大约3~4天得到初始的细菌纤维素凝胶。随后用0.1mol/l的naoh水溶液于80℃下纯化细菌纤维素凝胶,最后用二次蒸馏水反复冲洗至中性。得到的膜用粉碎机粉碎之后,经冷冻干燥得到高纯度的细菌纤维素粉末。随后,将10g细菌纤维素粉末分散在100ml质量分数为50%的浓硫酸溶液中,并强力搅拌。在45℃下反应5小时后,采用去离子水将反应溶液稀释5倍以终止水解反应。然后,在磁力搅拌作用下加入8ml质量分数为30%过氧化氢溶液对反应液进行氧化和漂白。30min后,反应液在9000r/min下离心分离得到沉淀。沉淀经反复水洗和超声处理后装入截留分子量为3500的透析袋中。在超纯水中透析1周后,产物经冷冻干燥制得平均水动力学粒径约为261nm,zeta电位为-30.6mv的bcns。
本实施例制得的bcns用于以下各实施例中。
实施例1
本实施例以caco3/gdl复合物为交联剂,庆大霉素为待负载的抗生素药物,在医用敷料的制备中,分别固定caco3与gdl的摩尔比为1:2,caco3中ca元素与sa中–cooh的摩尔比为0.36。
将5gsa溶解在200ml蒸馏水中,再加入50ml无水乙醇混合均匀。然后在上述混合液中加入1.72g(30%海藻酸钠糖醛酸单体摩尔量)高碘酸钠后,在室温下避光电动搅拌24h。向上述反应液中加入10ml乙二醇,避光磁力搅拌2h以终止反应。用5g氯化钠和800ml无水乙醇沉淀终止反应后的溶液。之后再将所得沉淀重新溶解在100ml的蒸馏水中,再以3g氯化钠和600ml无水乙醇沉淀该溶液。如此重复沉淀3次后,将最终所得溶液装入截留分子量为3500的透析袋中,透析5天,后经冷冻干燥就得到了干燥的高碘酸钠氧化海藻酸钠衍生物。然后将2g理论氧化度为30%的高碘酸钠氧化海藻酸钠衍生物溶解在120ml蒸馏水中。然后,将1.53g的正己胺溶解于10ml甲醇中。将溶有正己胺的甲醇溶液加入高碘酸钠氧化海藻酸钠衍生物溶液中,反应1h后,再加入0.95g氰基硼氢化钠,于室温下搅拌反应24h。将所得反应液装入截留分子量为8000的透析袋中,透析3天,后经冷冻干燥就得到了海藻酸胺化衍生物(raoa)。
将0.125gbcns、136.4mgcaco3纳米粉和50mg庆大霉素抗生素药物加入50ml质量分数为1.5%的raoa溶液中,在超声搅拌的作用下使其混合均匀。然后在快速搅拌作用下,加入485.8mggdl引发离子交联。所得凝胶置于0.01mol/lcacl2水溶液中浸泡冲洗3次,以除去未参与反应的化合物和杂质,最后所得复合凝胶经冷冻干燥后,浸入质量分数为0.5%的cs溶液中,孵化30min后取出,在去离子水中浸泡冲洗多次以除去未覆积的cs,直至洗液的ph值达到中性为止。清洗后的凝胶经冷冻干燥后浸入质量分数为1%的ha溶液中,同样孵化30min后取出,在去离子水中浸泡并反复冲洗。最后将清洗干净的复合凝胶投入2wt%京尼平溶液中孵育过夜。然后取出,用超纯水反复清洗,之后产物经冷冻干燥即得raoa/bcns复合凝胶功能性药物缓释医用敷料。
本实施例所得医用敷料能够显示出规整的3d形貌,且空隙大小分布均匀。其磷酸缓冲液中完全溶解时间为92天。l929细胞在该医用敷料上的存活率高达98.5%,且能够表现出较好的增殖效果。其抗菌效能如表1所示,对atccno.8739大肠杆菌和atccno.6538金黄色葡萄球菌的抑菌率分别为99.2%和98.8%。释药实验表明,该医用敷料在3天时间内能够持续缓慢地释放大约46%的庆大霉素抗生素药物,可以有效地减少换药次数。
实施例2
本实施例以caco3/gdl复合物为交联剂,氧氟沙星为待负载的抗生素药物,在医用敷料的制备中,分别固定caco3与gdl的摩尔比为1:2,caco3中ca元素与sa中–cooh的摩尔比为0.18。
将5gsa溶解在200ml蒸馏水中,再加入50ml无水乙醇混合均匀。然后在上述混合液中加入1.72g高碘酸钠后,在室温下避光电动搅拌24h。向上述反应液中加入10ml乙二醇,避光磁力搅拌2h以终止反应。用5g氯化钠和800ml无水乙醇沉淀终止反应后的溶液。之后再将所得沉淀重新溶解在100ml的蒸馏水中,再以3g氯化钠和600ml无水乙醇沉淀该溶液。如此重复沉淀3次后,将最终所得溶液装入截留分子量为3500的透析袋中,透析5天,后经冷冻干燥就得到了干燥的高碘酸钠氧化海藻酸钠衍生物。然后将2g理论氧化度为30%的高碘酸钠氧化海藻酸钠衍生物溶解在120ml蒸馏水中。然后,将1.96g的正辛胺溶解于10ml甲醇中。将溶有正辛胺的甲醇溶液加入高碘酸钠氧化海藻酸钠衍生物溶液中,反应1h后,再加入0.95g氰基硼氢化钠,于室温下搅拌反应24h。将所得反应液装入截留分子量为8000的透析袋中,透析3天,后经冷冻干燥就得到了海藻酸胺化衍生物(raoa)。
将0.375gbcns、90.9mgcaco3纳米粉和10mg氧氟沙星抗生素药物加入50ml质量分数为2.0%的raoa溶液中,在超声搅拌的作用下使其混合均匀。然后在快速搅拌作用下,加入323.8mggdl引发离子交联。所得凝胶置于0.01mol/lcacl2水溶液中浸泡冲洗3次,以除去未参与反应的化合物和杂质,最后所得复合凝胶经冷冻干燥后,浸入质量分数为1.5%的cs溶液中,孵化30min后取出,在去离子水中浸泡冲洗多次以除去未覆积的cs,直至洗液的ph值达到中性为止。清洗后的凝胶经冷冻干燥后浸入质量分数为3%的ha溶液中,同样孵化30min后取出,在去离子水中浸泡并反复冲洗。最后将清洗干净的复合凝胶投入2.5wt%戊二醛溶液中孵育过夜。然后取出,用超纯水反复清洗,之后产物经冷冻干燥即得raoa/bcns复合凝胶功能性药物缓释医用敷料。
本实施例所得医用敷料能够显示出规整的3d形貌,且空隙大小分布均匀。其磷酸缓冲液中完全溶解时间为81天。l929细胞在该医用敷料上的存活率高达99.1%,且能够表现出较好的增殖效果。其对atccno.8739大肠杆菌和atccno.6538金黄色葡萄球菌的抑菌率分别为97.6%和97.2%。释药实验表明,该医用敷料在3天时间内能够持续缓慢地释放大约65%的氧氟沙星抗生素药物,可以有效地减少换药次数。
实施例3
本实施例以caco3/gdl复合物为交联剂,氧氟沙星为待负载的抗生素药物,在医用敷料的制备中,分别固定caco3与gdl的摩尔比为1:2,caco3中ca元素与sa中–cooh的摩尔比为0.54。
将5gsa溶解在200ml蒸馏水中,再加入50ml无水乙醇混合均匀。然后在上述混合液中加入1.72g高碘酸钠后,在室温下避光电动搅拌24h。向上述反应液中加入10ml乙二醇,避光磁力搅拌2h以终止反应。用5g氯化钠和800ml无水乙醇沉淀终止反应后的溶液。之后再将所得沉淀重新溶解在100ml的蒸馏水中,再以3g氯化钠和600ml无水乙醇沉淀该溶液。如此重复沉淀3次后,将最终所得溶液装入截留分子量为3500的透析袋中,透析5天,后经冷冻干燥就得到了干燥的高碘酸钠氧化海藻酸钠衍生物。然后将2g理论氧化度为30%的高碘酸钠氧化海藻酸钠衍生物溶解在120ml蒸馏水中。然后,将2.38g的正癸胺溶解于10ml甲醇中。将溶有正癸胺的甲醇溶液加入高碘酸钠氧化海藻酸钠衍生物溶液中,反应1h后,再加入0.95g氰基硼氢化钠,于室温下搅拌反应24h。将所得反应液装入截留分子量为8000的透析袋中,透析3天,后经冷冻干燥就得到了海藻酸胺化衍生物(raoa)。
将0.5gbcns、409.1mgcaco3纳米粉和30mg氧氟沙星抗生素药物加入50ml质量分数为3.0%的raoa溶液中,在超声搅拌的作用下使其混合均匀。然后在快速搅拌作用下,加入1457.3mggdl引发离子交联。所得凝胶置于0.01mol/lcacl2水溶液中浸泡冲洗3次,以除去未参与反应的化合物和杂质,最后所得复合凝胶经冷冻干燥后,浸入质量分数为1.0%的cs溶液中,孵化30min后取出,在去离子水中浸泡冲洗多次以除去未覆积的cs,直至洗液的ph值达到中性为止。清洗后的凝胶经冷冻干燥后浸入质量分数为2.5%的ha溶液中,同样孵化30min后取出,在去离子水中浸泡并反复冲洗。最后将清洗干净的复合凝胶投入2.0wt%京尼平溶液中孵育过夜。然后取出,用超纯水反复清洗,之后产物经冷冻干燥即得raoa/bcns复合凝胶功能性药物缓释医用敷料。
本实施例所得医用敷料能够显示出规整的3d形貌,且空隙大小分布均匀。其磷酸缓冲液中完全溶解时间为129天。l929细胞在该医用敷料上的存活率高达98.9%,且能够表现出较好的增殖效果。其对atccno.8739大肠杆菌和atccno.6538金黄色葡萄球菌的抑菌率分别为98.7%和98.4%。释药实验表明,该医用敷料在3天时间内能够持续缓慢地释放大约39%的氧氟沙星抗生素药物,可以有效地减少换药次数。
实施例4
本实施例以caco3/gdl复合物为交联剂,庆大霉素为待负载的抗生素药物,在医用敷料的制备中,分别固定caco3与gdl的摩尔比为1:2,caco3中ca元素与sa中–cooh的摩尔比为0.36。
将5gsa溶解在200ml蒸馏水中,再加入50ml无水乙醇混合均匀。然后在上述混合液中加入1.72g高碘酸钠后,在室温下避光电动搅拌24h。向上述反应液中加入10ml乙二醇,避光磁力搅拌2h以终止反应。用5g氯化钠和800ml无水乙醇沉淀终止反应后的溶液。之后再将所得沉淀重新溶解在100ml的蒸馏水中,再以3g氯化钠和600ml无水乙醇沉淀该溶液。如此重复沉淀3次后,将最终所得溶液装入截留分子量为3500的透析袋中,透析5天,后经冷冻干燥就得到了干燥的高碘酸钠氧化海藻酸钠衍生物。然后将2g理论氧化度为30%的高碘酸钠氧化海藻酸钠衍生物溶解在120ml蒸馏水中。然后,将1.96g的正辛胺溶解于10ml甲醇中。将溶有正辛胺的甲醇溶液加入高碘酸钠氧化海藻酸钠衍生物溶液中,反应1h后,再加入0.95g氰基硼氢化钠,于室温下搅拌反应24h。将所得反应液装入截留分子量为8000的透析袋中,透析3天,后经冷冻干燥就得到了海藻酸胺化衍生物(raoa)。
将0.5gbcns、181.8mgcaco3纳米粉和50mg庆大霉素抗生素药物加入50ml质量分数为2.0%的raoa溶液中,在超声搅拌的作用下使其混合均匀。然后在快速搅拌作用下,加入647.7mggdl引发离子交联。所得凝胶置于0.01mol/lcacl2水溶液中浸泡冲洗3次,以除去未参与反应的化合物和杂质,最后所得复合凝胶经冷冻干燥后,浸入质量分数为1.5%的cs溶液中,孵化30min后取出,在去离子水中浸泡冲洗多次以除去未覆积的cs,直至洗液的ph值达到中性为止。清洗后的凝胶经冷冻干燥后浸入质量分数为3.0%的ha溶液中,同样孵化30min后取出,在去离子水中浸泡并反复冲洗。最后将清洗干净的复合凝胶投入分别含有10mmol/l的edc和10mmol/l的nhs混合交联剂中孵育过夜。然后取出,用超纯水反复清洗,之后产物经冷冻干燥即得raoa/bcns复合凝胶功能性药物缓释医用敷料。
本实施例所得医用敷料能够显示出规整的3d形貌,且空隙大小分布均匀。其磷酸缓冲液中完全溶解时间为103天。l929细胞在该医用敷料上的存活率高达98.8%,且能够表现出较好的增殖效果。其对atccno.8739大肠杆菌和atccno.6538金黄色葡萄球菌的抑菌率分别为99.0%和98.3%。释药实验表明,该医用敷料在3天时间内能够持续缓慢地释放大约51%的庆大霉素抗生素药物,可以有效地减少换药次数。
效果验证试验1
采用本发明制备的医用敷料对20例小鼠创面进行临床观察治疗,试验小鼠平均年龄24个月,人为创伤面积为20mm×20mm,创伤部分为小鼠背部,且均经过麻醉和消毒处理。20例小鼠均采用本发明实施例1制得的raoa/bcns复合凝胶功能性药物缓释医用敷料覆盖创面。伤后3天内,每天于09:00时更换创面敷料。创伤3天后,每2天于09:00时更换创面敷料,直至创面愈合。在整个换药期间进行创面观察。用药3天后组织液不再渗出,1周后创面无瘢痕,红肿消失。用药过程中未发现创面感染,无黏连。创面平均7天愈合,愈合后不留残余创面。上述临床实验结果表明,本发明制备的医用敷料具有良好的抗菌性、促愈合能力且无黏连。而且抗生素药物的持续释放,可以有效地减少换药次数,增强愈合效果。
效果验证试验2生物相容性试验
下述实施例中,采用小鼠成纤维l929细胞来评价raoa/bcns复合凝胶功能性药物缓释医用敷料的生物相容性。其细胞培养液为90%dmem,另外补加10%胎牛血清、100u/ml青霉素和100μg/ml链霉素。应用于细胞培养的raoa/bcns复合凝胶功能性药物缓释医用敷料,使用钴60射线灭菌,照射强度为8kgy。raoa/bcns复合凝胶功能性药物缓释医用敷料在接种细胞前先在细胞培养基中浸泡12小时以上。将复苏的细胞在聚苯乙烯细胞培养皿中传2代后采用胰蛋白酶消化并收集。细胞经计数后以每孔5×104的密度接种到24孔组织培养板的医用敷料上,同时将同样的细胞接种到无医用敷料的组织培养板上作为空白对照。补充细胞培养液,使每孔的培养基总量达到500μl。随后将组织培养板放入含5%co2的培养箱内,于37℃条件下培养,培养液每2天更换一次。通过细胞增殖-毒性检测试剂盒(cellcountingkit-8,cck-8)考察细胞在该医用敷料上的活力和增殖情况。同时,以革兰氏阴性菌atccno.8739大肠杆菌和革兰氏阳性菌atccno.6538金黄色葡萄球菌作为测试菌种,考察其抗菌活性。
对比例1
依据专利cn106637504a公布的方法制备得到海藻酸钙纤维敷料。在磷酸缓冲液中,其凝胶完全溶解时间为0.5天。l929细胞在该纤维敷料上的存活率高达95.3%,且能够表现出较好的增殖效果。其抗菌效能如表1所示,对atccno.8739大肠杆菌和atccno.6538金黄色葡萄球菌的抑菌率分别为87.7%和84.9%。
对比例2
准确称取1.0gsa溶于50ml去离子水中,于电力搅拌作用下制得sa溶液。随后依次将0.25gbcns、181.8mgcaco3纳米粉和25mg氧氟沙星抗生素药物加入该sa溶液中,在超声搅拌的作用下使其混合均匀。然后在快速搅拌作用下,加入647.7mggdl引发离子交联。所得凝胶置于0.01mol/lcacl2水溶液中浸泡冲洗3次,以除去未参与反应的化合物和杂质。最后所得复合凝胶经冷冻干燥(先在冰箱中于-18℃下预冻30min,后转入冷阱中于-50~60℃下冷冻5~6小时)后,浸入质量分数为1%的cs溶液中,孵化30min后取出,在去离子水中浸泡冲洗多次以除去未覆积的cs,直至洗液的ph值达到中性为止。清洗后的凝胶经冷冻干燥后浸入质量分数为2%的ha溶液中,同样孵化30min后取出,在去离子水中浸泡并反复冲洗。最后将清洗干净的复合凝胶投入分别含有10mmol/l的edc和10mmol/l的nhs混合交联剂中孵育过夜。然后取出,用超纯水反复清洗,之后产物经冷冻干燥即得sa/bcns复合凝胶功能性药物缓释医用敷料。所得医用敷料展现出规整的3d形貌,且空隙大小分布均匀。其磷酸缓冲液中完全溶解时间为57天。l929细胞在该医用敷料上的存活率高达97.4%,且能够表现出较好的增殖效果。其抗菌效能如表1所示,对atccno.8739大肠杆菌和atccno.6538金黄色葡萄球菌的抑菌率分别为94.1%和93.7%。释药实验表明,该医用敷料存在突释现象,在24小时内快速释放大约90%的氧氟沙星抗生素药物。
表1raoa/bcns复合凝胶功能性药物缓释医用敷料、sa/bcns复合凝胶功能性药物缓释医用敷料和单纯海藻酸钙纤维敷料的抗菌效能(p<0.001)
本发明所述的raoa/bcns复合凝胶构筑的功能性药物缓释医用敷料,以小鼠成纤维细胞(l929)作为模型细胞,通过细胞增殖-毒性检测试剂盒(cellcountingkit-8,cck-8)考察细胞在该医用敷料上的活力和增殖情况。
图4为l929细胞在raoa/bcns复合凝胶功能性药物缓释医用敷料上分别培养2天和5天后的细胞增殖情况。*代表p<0.05,表示有显著性差异。从图4中可以看出,l929细胞在raoa/bcns复合凝胶功能性药物缓释医用敷料上表现出较好的增殖能力,而且其增殖活力均高于对照组,说明l929细胞能够在raoa/bcns复合凝胶功能性药物缓释医用敷料的3d孔隙结构中生长。随着bcns添加量的升高,细胞的增殖活力均先升高后降低。当bcns添加量为0.5%~0.75%(w/v)时,细胞在复合医用敷料上显示出明显的增殖效果。
效果验证试验3
本发明制得的海藻酸胺化衍生物/细菌纤维素纳米晶复合凝胶构筑的功能性药物缓释医用敷料经刀片切割和喷金处理后,即可在扫描电镜(sem)下观察复合凝胶的孔隙结构形貌。
以caco3/gdl为交联体系,bcns为补强剂,通过raoa的内源交联制备的负载氧氟沙星的raoa/bcns复合凝胶功能性药物缓释医用敷料的实物图和剖切面扫描电镜图见图2。图2中的(a)为未干燥raoa/bcns复合凝胶功能性药物缓释医用敷料、(b)干燥raoa/bcns复合凝胶功能性药物缓释医用敷料和(c)raoa/bcns复合凝胶功能性药物缓释医用敷料在层层组装过程中的实物图;(d)~(i)为不同bcns添加量的raoa/bcns复合凝胶功能性药物缓释医用敷料扫描电镜图:(d)0.25%、(e)0.4%、(f)0.5%、(g)0.75%、(h)0.9%和(i)1.0%。
从图2中可以看出,raoa/bcns复合凝胶功能性药物缓释医用敷料显示规整的3d形貌。由于bcns较小的尺寸和良好的界面性能使其均匀地分散在凝胶基体中,从而使raoa/bcns复合凝胶功能性药物缓释医用敷料呈现均匀的淡蓝色光泽。在冷冻干燥过程中由于发生了相分离,使raoa/bcns复合凝胶功能性药物缓释医用敷料内部产生了均匀的孔隙结构,使其表现出较好的透气性。随着bcns含量的增加,raoa/bcns复合凝胶功能性药物缓释医用敷料的孔隙结构逐渐减小,孔径由350μm降至180μm左右。
效果验证试验4
raoa/bcns复合凝胶功能性药物缓释医用敷料的抗压强度采用微机控制电子万能材料试验机(ctm8050,协强仪器制造(上海)有限公司)进行测试。测试样品的底面直径为20mm,高10mm。测试时,负荷垂直于样品表面纵向压缩,压缩速率为5mm/min,当材料压碎或其应变大于60%时停止。在仪器记录的应力-应变曲线上,其线性最高点为该样品的抗压强度。每组样品平行进行5次试验,取其平均值。添加不同bcns条件下raoa/bcns复合凝胶功能性药物缓释医用敷料的抗压强度结果见图3。
由图3可知,随着bcns的含量的升高,其机械性能逐步升高。由于bcns的纤维网络结构和界面相互作用使raoa/bcns复合凝胶功能性药物缓释医用敷料的孔径减小,导致材料的机械性能增强。
以上所述的实施例只是本发明的一种较佳的方案,并非对本发明作任何形式上的限制,在不超出权利要求所记载的技术方案的前提下还有其它的变体及改型。
- 还没有人留言评论。精彩留言会获得点赞!