一种萃取剂及其制备方法与应用与流程
本发明属于稀土富集领域,尤其涉及一种萃取剂及其制备方法与应用。
背景技术:
由于稀土的独特性质及其在诸如发光,电子和磁性等高科技领域的应用,稀土行业在过去几十年中发展迅速。中国的稀土资源丰富,年产量占世界总产量的90%以上,特别是离子吸附型稀土矿,主要分布在我国江西、广东、湖南、广西、福建等地,研究发现,在离子吸附型稀土矿物中发现的中重稀土比其它富轻稀土矿物多十倍以上。现有技术中,通常利用硫酸铵作为浸出液从离子吸附矿物中回收稀土;然而,该浸出方法会造成水体中的氨氮污染,导致水体富营养化,降低水体中的生物多样性,而且浸出过程中流失的钙、镁等其他物质也会影响水体的植物生长。
离子吸附型矿物中稀土的总相比一般为0.05-0.3%,所以稀土的回收过程中必不可少的还需要对浸出液中浓度极低的稀土进行富集。现有技术中,经常使用碳酸氢铵或草酸沉淀来富集和回收稀土,但该过程稀土的回收率较低,且需要消耗大量的化学试剂,产生大量的氨氮废水或草酸废水,容易造成水体环境污染。据报道,当开采一吨稀土元素时,消耗3.5吨碳酸氢铵,产生1,000-1,200立方米的氨氮废水,而沉淀后的草酸废水由于其毒性和水溶性难以处理,其对环境有相当大的危害。此外,使用碳酸氢铵或草酸得到的稀土颗粒的尺寸极其细小,固液分离难度大。
技术实现要素:
因此,本发明针对现有技术中存在的缺点和不足,提供一种萃取剂及其制备方法与应用。
本发明提供一种萃取剂,所述萃取剂包括式(Ⅰ)结构化合物:
其中,所述式(Ⅰ)结构的化合物至少包括两个基团,R独立地选取代的碳原子数为3~12的直链烷基、未取代的碳原子数为3~12的直链烷基、取代的碳原子数为3~12的支链烷基、未取代的碳原子数为3~12的支链烷基、取代的芳基或未取代的芳基。
优选地,所述R独立的选自未取代的碳原子数为6的直链烷基、未取代的碳原子数为6的支链烷基。
本发明提供了上述萃取剂的制备方法,包括如下步骤:
(1)苯二酚类化合物和卤代烷基羧酸酯在溶剂中反应,得到粗酯;
(2)将粗酯溶解于无水乙醇、四氢呋喃,加入含有氢氧化物的乙醇溶液,得到反应混合物;
(3)除去反应混合物溶剂后,将残余物溶于水中,进行酸化,得到萃取剂。
优选地,所述(1)步骤包括:
将氢化钠溶于N,N-二甲基甲酰胺,加入对苯二酚、邻苯二酚或间苯二酚的溶液,反应得到苯酚钠溶液;
在苯酚钠溶液加入2-溴辛酸甲酯的溶液,设定温度下搅拌,得到混合物;
采用环己烷萃取,减压浓缩有机相,得到粗酯。
优选地,所述(3)还包括,
用1:1盐酸水溶液将水相的pH调节至约2-3,用乙酸乙酯萃取并用水洗涤后,收集有机相并减压浓缩,得到苯氧基二羧酸;
将粗酸用石油醚洗涤三次以上,并在正己烷中重结晶纯化以除去杂质。
优选地,苯二酚化合物与卤代烷基羧酸酯的摩尔比是(0.3~0.5):1。
优选地,无水乙醇:四氢呋喃的体积比为(1~2):1。
本发明还提供了利用上述萃取剂从硫酸镁浸出液中选择性富集稀土的方法,包括以下步骤:
(1)通过碱性化合物皂化上述萃取剂或上述制备方法制得的萃取剂,得到皂化萃取剂;
(2)将(1)制得的皂化萃取剂加入离子吸附型稀土矿物的MgSO4浸出液,充分生成沉淀物后,液/固离心分离后,得到稀土羧酸络合物及萃余液。
优选地,所述方法还包括:
采用酸液对所述稀土羧酸络合物进行洗脱,得到富稀土的溶液和萃取剂沉淀物。
优选地,所述碱性化合物包括氨水、氢氧化钠及氢氧化钾中的一种或多种。
优选地,所述固相萃取剂与所述稀土溶液中稀土元素的摩尔比为(1.5~2):1。
本发明实施例提出了一种利用苯氧羧酸衍生物作为萃取剂的新型稀土富集方法。和传统的液液萃取相比,该萃取-沉淀法至少包括如下有益效果:
本发明合成了烷基苯氧基羧酸衍生物,并将其作为固体萃取剂,从离子吸附矿物的MgSO4浸出液中富集稀土,利用本发明合成的萃取剂对来自MgSO4浸出溶液的RE具有良好的选择性,在液/固相比为450/1条件下,能够获得良好的沉淀率,稀土沉淀尺寸较大,固液分离容易,且无挥发性稀释剂的使用,萃余液及羧酸萃取剂均能够回收再循环使用,符合中国国家废水排放标准。
附图说明
为了更清楚地说明本发明实施例或现有技术中的技术方案,下面将对实施例或现有技术描述中所需要使用的附图作简单地介绍,显而易见地,下面描述中的附图仅仅是本发明的实施例,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据提供的附图获得其他的附图。
图1是萃取剂和再生萃取剂的IR谱图。
图2是PPBOA为的1H和13C NMR光谱图。
图3是OPBOA为的1H和13C NMR光谱图。
图4是MPBOA为的1H和13C NMR光谱图。
图5不同萃取剂沉淀稀土的效率对比图。
图6是稀土固体沉淀物的形貌和尺寸图。
图7是萃余液和新制浸出试剂对离子吸附型稀土矿的浸出曲线对比图。
图8是从硫酸镁浸出液中富集稀土的流程图。
具体实施方式
下面对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
本发明提供一种萃取剂,所述萃取剂包括式(Ⅰ)结构化合物:
其中,所述式(Ⅰ)结构的化合物至少包括两个基团,进一步地R独立地选自取代的碳原子数为3~12的直链烷基、未取代的碳原子数为3~12的直链烷基、取代的碳原子数为3~12的支链烷基、未取代的碳原子数为3~12的支链烷基、取代的芳基或未取代的芳基。
优选地,所述R独立的选自未取代的碳原子数为6的直链烷基、未取代的碳原子数为6的支链烷基,具体的为2,2'-(1,4-亚苯基双(氧)基)二辛酸,分子结构如式(II);2,2'-(1,2-亚苯基双(氧)基)二辛酸,如式(III);2,2'-(1,3-亚苯基双(氧)基)二辛酸),如式(iv),其中式(Ⅰ)的R为C6H13。
本发明提供了上述萃取剂的制备方法,包括如下步骤:
(1)苯二酚类化合物和卤代烷基羧酸酯在溶剂中反应,得到粗酯;
(2)将粗酯溶解于无水乙醇、四氢呋喃,加入含有氢氧化物的乙醇溶液,得到反应混合物;
(3)除去反应混合物溶剂后,将残余物溶于水中,进行酸化,得到萃取剂。
在本发明提供的上述萃取剂制备方法中,首先将所述苯二酚类化合物和卤代烷基羧酸酯在溶剂中混合。其中,所述卤代烷基羧酸酯中的烷基可以独立地选自取代的碳原子数为3~12的直链烷基、未取代的碳原子数为3~12的直链烷基、取代的碳原子数为3~12的支链烷基、未取代的碳原子数为3~12的支链烷基,优选地选自取代的碳原子数为3的直链烷基、取代的碳原子数为4的直链烷基、取代的碳原子数为5的直链烷基、取代的碳原子数为6的直链烷基、取代的碳原子数为7的直链烷基、取代的碳原子数为8的直链烷基、取代的碳原子数为9的直链烷基、取代的碳原子数为10的直链烷基、取代的碳原子数为11的直链烷基、取代的碳原子数为12的直链烷基、未取代的碳原子数为3的直链烷基、未取代的碳原子数为4的直链烷基、未取代的碳原子数为5的直链烷基、未取代的碳原子数为6的直链烷基、未取代的碳原子数为7的直链烷基、未取代的碳原子数为8的直链烷基、未取代的碳原子数为9的直链烷基、未取代的碳原子数为10的直链烷基、未取代的碳原子数为11的直链烷基、未取代的碳原子数为12的直链烷基、取代的碳原子数为3的支链烷基、取代的碳原子数为4的支链烷基、取代的碳原子数为5的支链烷基、取代的碳原子数为6的支链烷基、取代的碳原子数为7的支链烷基、取代的碳原子数为8的支链烷基、取代的碳原子数为9的支链烷基、取代的碳原子数为10的支链烷基、取代的碳原子数为11的支链烷基、取代的碳原子数为12的支链烷基、未取代的碳原子数为3的支链烷基、未取代的碳原子数为4的支链烷基、未取代的碳原子数为5的支链烷基、未取代的碳原子数为6的支链烷基、未取代的碳原子数为7的支链烷基、未取代的碳原子数为8的支链烷基、未取代的碳原子数为9的支链烷基、未取代的碳原子数为10的支链烷基、未取代的碳原子数为11的支链烷基或未取代的碳原子数为12的支链烷基。
所述(1)步骤包括:将氢化钠溶于N,N-二甲基甲酰胺,加入对苯二酚、邻苯二酚或间苯二酚的溶液,反应得到苯酚钠溶液;在苯酚钠溶液加入2-溴辛酸甲酯的溶液,设定温度下搅拌,得到混合物;采用环己烷萃取,减压浓缩有机相,得到粗酯。
优选地,所述(3)还包括,用盐酸水溶液将水相的pH调节至约2-3,用乙酸乙酯萃取并用水洗涤后,收集有机相并减压浓缩,得到苯氧基二羧酸;将粗酸用石油醚洗涤三次以上,并在正己烷中重结晶纯化以除去杂质。
优选地,苯二酚化合物与卤代烷基羧酸酯的摩尔比是(0.3~0.5):1。
优选地,无水乙醇:四氢呋喃的体积比为(1~2):1。
本发明还提供了利用上述萃取剂从硫酸镁浸出液中选择性富集稀土的方法,包括以下步骤:
(1)通过碱性化合物皂化上述萃取剂或上述制备方法制得的萃取剂,得到皂化萃取剂;
(2)将(1)制得的皂化萃取剂加入离子吸附型稀土矿物的MgSO4浸出液,充分生成沉淀物后,液/固离心分离后,得到稀土羧酸络合物及萃余液。
在(1)中,通过酸碱中和反应(方程式i)的皂化过程去除质子和破坏萃取剂中的二聚体,提高酸性萃取剂的萃取率和选择性。
NaOH+H2A→Na2A+H2O (i)
其中,H2A为上述方法制备的萃取剂(固体羧酸),NaOH为碱性化合物,所述碱性化合物还可以包括氨水、氢氧化钾等碱性溶液。
在(2)中,沉淀反应可写成以下式(ii):
RE3++A2-→RE2A3↓ (ii)
在上述富集方法中,不同皂化程度下的萃取剂与稀土溶液反应,稀土离子沉淀率(萃取剂萃取性能的指标)不同,随着沉淀过程中皂化度的增加,PPBOA,OPBOA和MPBOA的水溶性略有增加;当皂化程度在0~80%的范围内增加,稀土与PPBOA,OPBOA和MPBOA的沉淀效率逐渐增加。由于未皂化的羧酸也具有一定的萃取效果,当皂化度低时,固体萃取剂中H+的减少的摩尔量(ΔH+)/生成固体络合物中稀土元素的摩尔量(ΔRE3+)的比率显着低于1.5;而随着皂化度的增加,ΔH+/ΔRE3+的比率逐渐接近1.5并且基本保持不变,即随着皂化程度增加,稀土离子沉淀率逐渐增加。
在(1)中,将H2A萃取剂应用碱皂化提高其萃取能力,再将MgSO4浸出液与皂化的萃取剂(Na2A)接触,产生大量的白色沉淀;固液分离后,可以得到白色稀土羧酸络合物(A-RE)和萃余液。使用萃取-沉淀系统,不仅可以获得高浓度的稀土溶液,固体羧酸萃取剂也可以恢复到其原始状态以便再循环。
优选地,所述方法还包括:采用酸液对所述稀土羧酸络合物进行洗脱,得到富稀土的溶液与萃取剂沉淀物。
固体萃合物在用浓HCl进行反萃后,通过IR表征再生的PPBOA,OPBOA和MPBOA,在萃取-沉淀过程之前和之后,在COOH中的C=O的伸缩振动的1701cm-1,1698cm-1和1709cm-1的特征峰是一致,如图1所示,表明PPBOA,OPBOA和MPBOA在再循环过程中的稳定性。与一次性使用的草酸和碳酸氢铵相比,可回收的苯氧基羧酸不仅环保,而且还节省成本。
优选地,所述固相萃取剂与所述稀土溶液中稀土元素的摩尔比为(1.5~2):1。
为更清楚起见,下面通过以下实施例进行详细说明。
实施例1
1、萃取剂2,2'-(1,4-亚苯基双(氧)基)二辛酸(PPBOA)的制备方法
(1)将1.80g(0.045mol)氢化钠,20mlN,N-二甲基甲酰胺(DMF)加入反应容器中。在室温下搅拌,在氩气(或者氮气)保护的同时缓慢加入对苯二酚的溶液(0.02mol,2.20g对苯二酚在20ml DMF中),得到苯酚钠溶液;缓慢加入2-溴辛酸甲酯(0.05mol,11.9g)的溶液,60℃下搅拌6小时;用环己烷萃取后,减压浓缩有机相,最后得到相应的粗酯。
其中,上述选用的试剂采用同类试剂替换也能够实现萃取剂的制备,例如:氢化钠也可以用其它碱替代,N,N-二甲基甲酰胺也可以采用N,N-二甲基乙酰胺;2-溴辛酸甲酯也可以用其它卤代羧酸酯替代,如2-溴辛酸乙酯,环己烷也可以用正己烷替代。
(2)2,2'-(1,4-亚苯基双(氧)基)二辛酸二甲酯(因为是粗酯,无法确定实际的质量,实验的时候用的质量是理论值,以防加的碱不够),无水乙醇(或者甲醇)(C2H5OH)(60ml)和四氢呋喃(THF)(VC2H5OH:VTHF,优选为1:1)加入反应溶剂搅拌;溶解后,室温下缓慢加入含有KOH(或者NaOH)(0.08mol,4.6448g)的60ml乙醇溶液,加热回流6小时。
(3)除去溶剂后,将残余物溶于水中,用1:1盐酸水溶液将水相的pH调节至约2-3。用乙酸乙酯萃取并用水洗涤后,收集有机相并减压浓缩,得到相应的粗酸;将粗酸用石油醚洗涤三次以上,并在正己烷中重结晶纯化以除去杂质,得到2,2'-(1,4-亚苯基双(氧)基)二辛酸(PPBOA)(如式II),作为萃取剂。
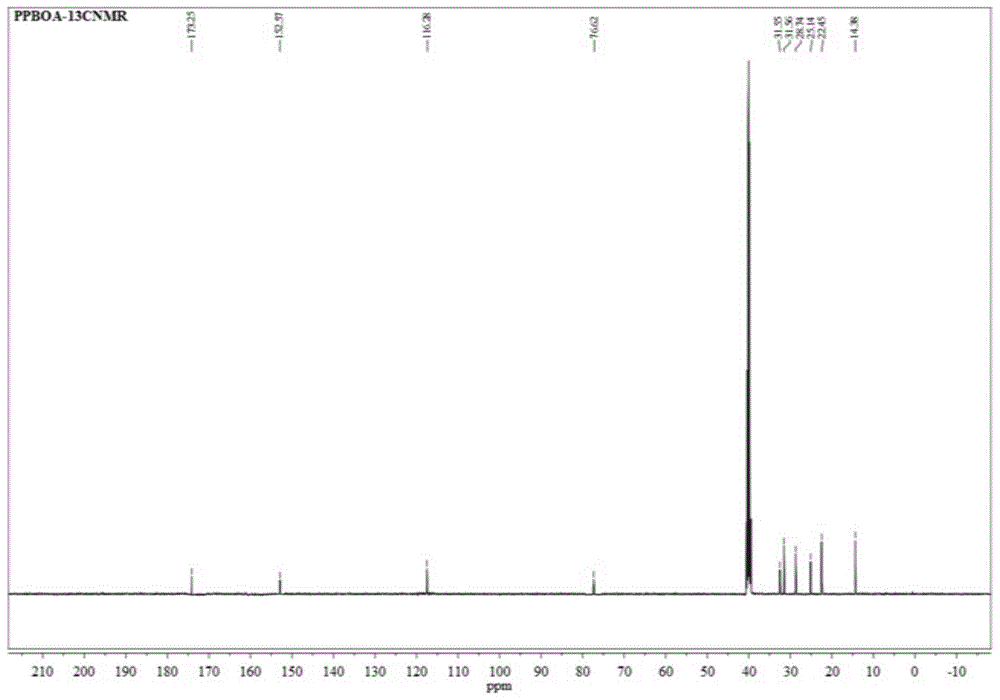
通过上述方法制得的PPBOA产率为63%,纯度>99%,熔点132℃,25℃下水溶性17.5ppm(紫外-可见光谱测出);1H NMR(DMSO-d6,ppm)δ12.92(s,2H),6.79(s,4H),4.55(t,J=6.2Hz,2H),1.80(m,4H),1.51–1.18(m,16H),0.86(t,J=6.9Hz,6H);13C NMR(DMSO-d6,ppm)δ173.25,152.57,116.28,76.62,32.61,31.55,28.76,25.14,22.45,14.38.通过AV III-500Bruker光谱仪获得1H光谱图(如图2-1)和13C NMR光谱图(如图2-2)。
2、从硫酸镁浸出液中富集稀土的方法
(1)制备硫酸镁浸出液
MgSO4浸出过程是一种离子交换的过程,可以用收缩芯模型来描述,其中内扩散控制浸出速度。浸出剂中的镁离子与矿物之间的离子交换过程的化学反应方程如下(式iii)。
(2)皂化萃取剂
将上述制得PPBOA与氢氧化钠反应,如式(i)。
(3)室温下萃取10分钟,通过使皂化的PPBOA萃取剂与浸出液接触形成固体复合物,如式(ii),通过在4000rpm下离心进行固液分离。
当使用20ml MgSO4浸出溶液(稀土浓度调节为0.392g/L)与皂化的萃取剂接触时,产生大量沉淀物(液/固相比=450/1),液/固离心分离后,ICP-OES测定PPBOA萃余液中稀土的浓度为0.0161g/L,相应萃取剂的稀土沉淀率达到95.1%。
实施例2
1、萃取剂2,2'-(1,2-亚苯基双(氧)基)二辛酸(OPBOA)的制备方法
将1.80g(0.045mol)氢化钠,20ml N,N-二甲基甲酰胺(DMF)加入装有冷凝及搅拌装置的反应容器中,在室温下搅拌,在氩气保护的同时缓慢加入邻苯二酚的溶液,得到苯酚钠溶液,缓慢加入2-溴辛酸甲酯(0.05mol,11.9g)的溶液,在75℃下搅拌6小时;用环己烷萃取后,减压浓缩有机相,最后得到相应的粗酯。
(2)2,2'-(1,2-亚苯基双(氧)基)二辛酸二甲酯,无水乙醇(C2H5OH)(60ml)和四氢呋喃(THF)(VC2H5OH:VTHF,1:1)加入反应容器中;完全溶解后,在室温下逐滴加入含有KOH(0.08mol,4.6448g)的60ml乙醇溶液,加热回流6小时。
(3)除去溶剂后,将残余物溶于水中,用1:1盐酸水溶液将水相的pH调节至约2-3。用乙酸乙酯萃取并用水洗涤后,收集有机相并减压浓缩,得到相应的粗酸;将粗酸用石油醚洗涤三次以上,并在正己烷中重结晶纯化以除去杂质,得到2,2'-(1,2-亚苯基双(氧)基)二辛酸(OPBOA)(如式III),作为萃取剂。
通过上述方法制得的OPBOA产率为51%(0.0102mol,4.02g).纯度>98%.1H NMR(DMSO-d6,ppm)δ12.81(s,2H),6.88(s,4H),4.69(t,J=6.0Hz,2H),1.85(m,4H),1.57–1.17(m,16H),0.87(t,J=6.8Hz,6H);13C NMR(DMSO-d6,ppm)δ173.19,148.44,122.38,116.94,77.27,32.94,31.67,29.03,25.13,22.5,14.38.熔点118℃.25℃下的水溶性29.0ppm。
通过AV III-500Bruker光谱仪获得1H光谱图(如图3-1)和13C NMR光谱图(如图3-2)。
2、从硫酸镁浸出液中富集稀土的方法
通过使皂化的OPBOA与浸出液反应形成固体复合物,如式(ii),在室温下萃取10分钟,通过在4000rpm下离心进行固液分离。
当使用20ml MgSO4浸出溶液(稀土浓度调节为0.392g/L)与皂化的OPBOA反应时,产生大量沉淀物(液/固相比=450/1),液/固离心分离后,ICP-OES测定OPBOA萃余液中稀土的浓度为0.0213g/L,相应萃取剂的稀土沉淀率达到93.5%。
实施例3
1、萃取剂2,2'-(1,3-亚苯基双(氧)基)二辛酸(PPBOA)的制备方法
(1)将1.80g(0.045mol)氢化钠,20ml N,N-二甲基甲酰胺(DMF)加入装有冷凝及搅拌装置的反应容器中;在氩气保护的同时缓慢加入间苯二酚的溶液,得到苯酚钠溶液,再缓慢加入2-溴辛酸甲酯(0.05mol,11.9g)的溶液;85℃下搅拌6小时;用环己烷萃取后,减压浓缩有机相,最后得到相应的粗酯。
(2)2,2'-(1,3-亚苯基双(氧)基)二辛酸二甲酯,无水乙醇(C2H5OH)(60ml)和四氢呋喃(THF)(VC2H5OH:VTHF,1:1)加入反应容器中;完全溶解后,在室温下缓慢加入含有KOH(0.08mol,4.6448g)的60ml乙醇溶液。将反应混合物在空气气氛下加热回流6小时。
(3)除去溶剂后,将残余物溶于水中,用1:1盐酸水溶液将水相的pH调节至约2-3。用乙酸乙酯萃取并用水洗涤后,收集有机相并减压浓缩,再将得到的粗酸用石油醚洗涤三次以上,并在正己烷中重结晶纯化以除去杂质,得到2,2'-(1,3-亚苯基双(氧)基)二辛酸)(MPBOA)(如式IV),作为萃取剂。
通过上述方法制得的MPBOA产率34%(0.0068mol,2.68g).纯度>98%.熔点115℃.25℃下的水溶性21.2ppm.1H NMR(DMSO-d6,ppm)δ12.98(s,2H),7.15(t,J=8.3Hz,1H),6.44(d,J=8.1Hz,2H),6.36(s,1H),4.64(t,J=6.2Hz,2H),1.82(m,4H),1.50–1.16(m,16H),0.87(t,J=6.8Hz,6H);13C NMR(DMSO-d6,ppm)δ172.98,159.37,130.44,107.69,102.68,75.92,32.52,31.54,28.74,25.12,22.45,14.38.通过AV III-500Bruker光谱仪获得1H光谱图(如图4-1)和13C NMR光谱图(如图4-2)。
2、从硫酸镁浸出液中富集稀土的方法
通过使皂化的MPBOA与浸出液反应形成固体复合物,如式(ii),在室温下萃取10分钟,通过在4000rpm下离心进行固液分离。
当使用20ml MgSO4浸出溶液(稀土浓度调节为0.392g/L)与皂化的MPBOA反应时,产生大量沉淀物(液/固相比=450/1),液/固离心分离后,ICP-OES测定MPBOA萃余液中稀土的浓度为0.0108g/L,相应萃取剂的稀土沉淀率为96.7%。
对比例1
利用对叔辛基苯氧基乙酸(POAA)从硫酸镁浸出液中富集稀土
对叔辛基苯氧基乙酸(POAA)作为具有良好性能的固体萃取剂,其制备方法可以参考:《Enrichment of trace rare earth elements from the leaching liquor of ion-absorption minerals using a solid complex centrifugal separation process》(Green Chem.2018,20,1998-2006);将其与PPBOA,OPBOA和MPBOA进行比较。
保持稀土浓度不变,随着镁浓度逐渐增加(Mg与RE的比例增加),稀土与POAA的沉淀效率显着降低,而与PPBOA,OPBOA和MPBOA的沉淀效率几乎保持不变(>98%),如图5所示,图5描绘了来自POAA,PPBOA,OPBOA和MPBOA的RE和Mg的沉淀效率,相比之下,POAA显示出较低的稀土沉淀效率和较高的Mg沉淀效率。很明显,POAA对RE的选择性显着低于其他三种新型沉淀剂。镁沉淀物一部分来自萃取,另一部分来自与RE的共沉淀;对于POAA系统,Mg沉淀物主要来自萃取。而对于PPBOA,OPBOA和MPBOA系统,Mg沉淀物主要来自共沉淀。在相同条件下,当使单一硫酸镁(MgSO4)溶液与萃取剂接触时,POAA可产生显着的沉淀,而其他三种XPBOA萃取剂则不能产生沉淀的实验可以证明这一点。由于PPBOA,OPBOA,MPBOA中的双羧基和POAA中的单羧基,PPBOA,OPBOA,MPBOA的沉淀能力,即136.3mg/g,131.1mg/g和143.5mg/g,明显高于POAA(98.15mg/g)。
由此可以得出,与以前的固体萃取剂POAA相比,在高浓度镁存在下,本发明制备的苯氧基二羧酸衍生物萃取剂显示出比POAA更高的沉淀能力和更好的稀土选择性。
对比例2
与通过现有技术中H2C2O4和NH4HCO3获得的稀土沉淀物的粒径的比较结果。
粒径对固液分离过程中稀土的回收率有显著的影响。为了进行比较,收集了PPBOA,OPBOA和MPBOA制备的沉淀物。图6表示固体沉淀物的形貌与尺寸,其中,(a)的萃取沉淀剂为PPBOA(NaOH皂化),(b)的萃取沉淀剂为OPBOA(NaOH皂化),(c)的萃取沉淀剂为MPBOA(NaOH皂化),(d)的萃取沉淀剂为PPBOA(NH3·H2O皂化),(e)的沉淀剂为H2C2O4,(f)沉淀剂为NH4HCO3。如图6所示,用NaOH皂化的PPBOA,OPBOA和MPBOA制备的沉淀与NH3·H2O皂化的PPBOA形貌非常相似,粒径范围在57-88μm;而与H2C2O4和NH4HCO3制备的沉淀物的粒径范围为3.6-5.5μm。
由此可以得出,通过本发明萃取剂获得的稀土沉淀物与现有技术中通过H2C2O4和NH4HCO3获得稀土沉淀物具有更大的粒径,更有助于稀土萃取络合物和水相的分离,提高生产效率。
对比例3
考虑到沉淀效应和环境污染问题(COD值),选择1500ml进料溶液和3.33gMPBOA作为沉淀剂进行循环实验。干燥沉淀后,得到3.58g白色固体。将固体置于含有磁力搅拌器的圆底烧瓶中,加入2ml浓盐酸(固/液相比约为1.8/1),加热至100℃实现完全剥离,获得高浓度的238.0g/L稀土溶液。
用MPBOA系统的萃余液和首次使用的MgSO4溶液进行浸出试验,验证萃余液的循环可行性。加入适量MgSO4后,萃余液中MgSO4的浓度为23.1g/L,新鲜MgSO4溶液的浓度为22.8g/L.整个浸出过程在3.2毫米直径的PMMA透明柱中进行,其中填充的离子吸附矿物质为250g,柱高为22厘米。由图7可知,首次使用的MgSO4溶液和再循环MgSO4溶液的浸出曲线是相似,表明在萃余液中存在极少量的萃取剂不会影响浸出过程,并且萃余液是可回收的;通过萃取剂从硫酸镁浸出液中富集稀土的流程如图8所示,固液分离后,可以得到白色稀土羧酸络合物(A-RE)和萃余液,并且固体羧酸萃取剂可以恢复到其原始状态以便再循环。
以上所述仅是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。
- 还没有人留言评论。精彩留言会获得点赞!