一种球形纳米羟基磷灰石颗粒的制备方法与流程
本发明属于生物医用材料技术领域,涉及一种新型的球形纳米羟基磷灰石颗粒的制备方法。
背景技术:
羟基磷灰石(hydroxyapatite,HAP)作为生理条件下最稳定的磷酸钙盐,是脊椎动物骨骼和牙齿的主要无机矿物成分。因具有良好的生物相容性和生物活性,被广泛的用于组织工程、骨的修复与替代和牙科材料等,已被证实介孔HAP能够通过骨传导机制促进新骨的生成而不引起任何毒性、炎症和排斥反应。此外,由于独特的化学成分和晶体结构,HAP对生物分子展示了高亲和力,如DNA和RNA,因此也被广泛的用于蛋白质、药物、基因、生长因子的控制释放载体。例如,作为药物载体,纳米级颗粒可以更快的通过细胞膜,保护治疗药物免受酶降解,调控药物的释放性能,还可以通过配位分子表面改性或功能化提高靶向性到达特定病变细胞或组织。
近年来,随着对纳米HAP研究的快速发展,研究人员对HAP的特性有了更深入的了解,已被证实HAP的物化性能主要取决于其形貌、粒径和结晶度。目前,国内外对于不同形貌羟基磷灰石及制备方法的研究较多。在现有检索到的文献中,Yang等采用水热法在柠檬酸辅助下合成出中空的HAP微球[Yang H,Hao L J,Zhao N R,et al.Hierarchical porous hydroxyapatite microsphere as drug delivery carrier.Crystengcomm,2013,15(29),5760-5763],但是所得到的微球是由尺寸在400nm左右的纳米棒或纳米片作为结构单元聚合而成,且合成过程要经高温高压,对设备要求较高。Lin等在未使用任何表面活性剂、模板支持和结构导向剂的前提下,利用简单的低温水热过程制备出了具有均匀的含碳磷灰石微球[Lin K L,Chang J,Zhu Y J,et al.A facile one-step surfactant-free and low-temperature hydrothermal method to prepare uniform 3D structured carbonated apatite flowers.Cryst Growth Des,2009,9(1),177-181],这些含碳磷灰石是由大量厚度约为75nm,宽200-1000nm,长度可达5μm的纳米板聚合而成,产物的结晶度较低。吕宇鹏等(CN1903706A)以四水硝酸钙(Ca(NO3)2·4H2O)和磷酸氢二铵((NH4)2HPO4)为原料,采用化学沉淀法制得羟基磷灰石料浆,然后将其稀释加入碳酸氢铵,搅拌均匀后进行喷雾处理,结合后续热处理,得到了由纳米晶粒组成的空心的羟基磷灰石微球,所制备的羟基磷灰石微球粒度均匀,表面积大,空心率高,但是结晶度较低,粒径较大。然而,到目前为止,具有规整形貌的球形纳米羟基磷灰石报道的却很少。特别是以碳量子点为核,羟基磷灰石对其包覆得到球形的颗粒还没有相关报道。
碳量子点(CQDs),作为近年来才被发现的新型零维纳米碳材料,其粒径小于10nm,具有稳定性高、生物相容性好而且有很强的可调控光致发光等特性。在碳量子点的表面含有大量的-OH和-COOH,为后续过程吸附其他前驱物或纳米粒子提供了良好的化学环境。
技术实现要素:
本发明的目的是提供一种球形纳米羟基磷灰石颗粒的制备方法,以CQDs作为中心核,诱导HAP沉积在其表面,从而避免HAP的无序生长得到规整形貌的球形HAP纳米颗粒。
本发明的目的是通过以下技术方案实现的:
一种球形纳米羟基磷灰石颗粒的制备方法,包括如下步骤:
(1)量取20ml浓度为0.0625~1.00mmol/L的CQDs溶液至锥形瓶中,调节溶液pH升至10,室温搅拌10~30min;
(2)逐滴加入1ml浓度为40~100mmol/L的CaCl2溶液,均匀搅拌20~40min;
(3)逐滴加入1ml浓度为24~60mmol/L的Na2HPO4·12H2O溶液,控制Ca/P摩尔比1.7,于室温下静置90~100h;
(4)制得的产物离心分离,弃去上层清液,下层产物分别用蒸馏水和无水乙醇洗3~5次,洗涤后的产品取出,在烘箱中干燥得到土黄色粉末;
(5)将粉末放入马弗炉由室温升温到500~600℃,在该温度下煅烧4~6h,除去碳量子点得到白色粉末即为HAP纳米颗粒。
本发明中,以蔗糖作为碳源,采用水热法制备碳量子点,具体步骤如下:量取5.0ml浓度为0.01mol/L的蔗糖溶液、1.0ml稀H2SO4溶液、44.0ml蒸馏水三者置于同一锥形瓶中,在磁力搅拌下使之混合均匀,然后将溶液转移至聚四氟乙烯高压反应釜中,于140℃反应8h,自然冷却至室温,得到CQDs溶液。
利用本发明的方法,以碳量子点作为有效的成核诱导剂,采用化学沉淀法,在室温下得到了球形的羟基磷灰石纳米颗粒。本发明所制备的羟基磷灰石呈准球形,具有良好的分散性,颗粒均匀,结晶度高。此外,制备方法简单,条件温和可控,成本较低。
附图说明
图1为本发明最佳条件下所得球形纳米羟基磷灰石的X射线衍射(XRD)图谱与羟基磷灰石的标准图谱;
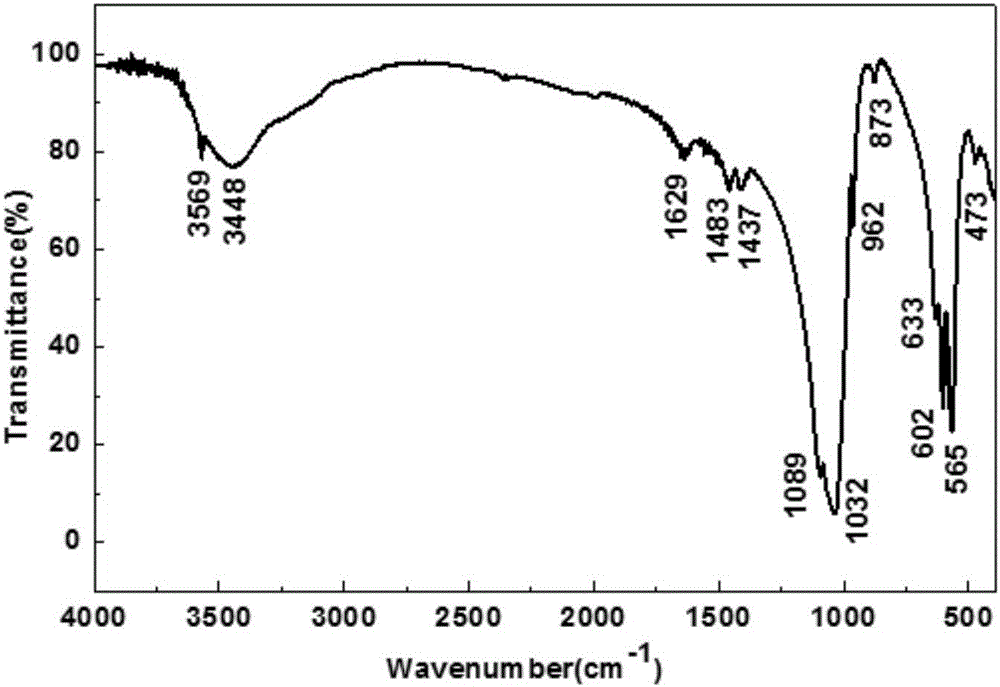
图2为本发明最佳条件下煅烧后羟基磷灰石的傅里叶变换红外光谱图(FTIR);
图3为不同碳量子点浓度对产物形貌的影响,a:0,b:0.0625mmol/L,c:0.250mmol/L,d:1.00mmol/L;
图4为CaCl2加入量对产物形貌的影响,a:40mmol/L,b:60mmol,c:80mmol/L,d:100mmol/L。
具体实施方式
下面结合附图对本发明的技术方案作进一步的说明,但并不局限于此,凡是对本发明技术方案进行修改或者等同替换,而不脱离本发明技术方案的精神和范围,均应涵盖在本发明的保护范围中。
本发明提供了一种球形纳米羟基磷灰石颗粒的制备方法,首先,蔗糖的稀溶液在水热处理下制备碳量子点,将其作为有效的成核诱导剂,其中硫酸作为碳化剂;然后用去离子水将其稀释,滴加氢氧化钠水溶液调节溶液pH值,采用无水氯化钙和磷酸氢二钠为原料,Ca2+通过静电引力吸附在碳量子点的表面形成一个“蛋-壳”结构;加入磷源后,PO43-与Ca2+反应在碳量子点的表面生成磷酸钙盐,经过奥斯特瓦尔德成熟,最后转化为更为稳定的HAP从而得到球形HAP纳米颗粒。具体步骤如下:
一、CQDs的制备
溶液的配制:称取0.343g蔗糖溶于100ml蒸馏水中,配置成0.01mol/L的蔗糖溶液;移液管量取0.10ml浓H2SO4用蒸馏水稀释至10ml,混合均匀。
碳量子点的制备:量取5.0ml上述蔗糖溶液、1.0ml稀H2SO4溶液、44.0ml蒸馏水三者置于同一锥形瓶中,在磁力搅拌下使之混合均匀,然后将溶液转移至聚四氟乙烯高压反应釜中,于140℃反应8h,自然冷却至室温,得到CQDs溶液。
二、纳米HAP的制备
溶液配制:称取0.053g无水CaCl2溶于6ml蒸馏水中,配置成80mmol/L的氯化钙溶液;同样称取0.103g Na2HPO4·12H2O溶于6ml蒸馏水中,Ca/P摩尔比1.7,配置成48mmol/L的磷酸氢二钠溶液;称取0.200g NaOH溶于5ml蒸馏水中,配置成1mol/L的氢氧化钠溶液;量取上述制备的CQDs溶液20ml到60ml蒸馏水中稀释,搅拌混合均匀,得到浓度为0.25mmol/L的碳量子点溶液。
纳米HAP颗粒的制备:量取上述稀释后的CQDs溶液20ml至锥形瓶中,搅拌中逐滴加入1mol/L的NaOH水溶液调节溶液pH升至10,室温搅拌20min,随后逐滴加入1ml CaCl2溶液,均匀搅拌30min,然后逐滴加入1ml Na2HPO4·12H2O溶液,于室温下静置96h。制得的产物在9000r/min下离心分离,弃去上层清液,下层产物分别用蒸馏水和无水乙醇洗3次,洗涤后的产品取出,在烘箱中100℃下干燥得到土黄色粉末。将粉末放入马弗炉由室温升温(2℃/min)到550℃,在该温度下煅烧5h,除去碳量子点得到白色粉末即HAP纳米颗粒。
图1为本实施例制备的样品的XRD图谱与HAP的标准图谱。对照HAP的标准图谱(JCPDS 09-0432)可知,煅烧后样品b的XRD图谱中各衍射峰都能与其一一对应,没有其他杂质峰的出现,说明所制备的样品中形成了HAP而不是其他的磷酸钙盐,HAP为六方晶系,属于P63/m空间群。位于2θ=26.73°,31.80°,33.57°,34.12°,39.86°,46.81°,48.16°和49.54°处的强衍射峰分别是HAP(002)、(211)、(300)、(202)、(310)、(222)、(213)和(004)晶面的衍射峰。此外,从未煅烧样品a的XRD图谱中可以看到各衍射峰较宽,峰强较弱;经过煅烧后,可以看出样品b的衍射峰基本没有重叠现象,变得较为尖锐,峰强明显增强,结晶度得到提高。
从图2中可以看出,3569cm-1和633cm-1处的吸收峰分别为OH-的伸缩振动峰和弯曲振动峰。四面体结构PO43-的特征吸收峰位于473cm-1、565cm-1、602cm-1、962cm-1、1032cm-1和1089cm-1处。473cm-1对应PO43-的γ2振动吸收峰,565cm-1、602cm-1对应PO43-的γ4振动吸收峰,962cm-1对应PO43-的γ1振动吸收峰,1032cm-1和1089cm-1对应PO43-的γ3振动吸收峰。另外,873cm-1以及1437cm-1和1483cm-1处的吸收峰为CO32-的吸收峰,这些峰的出现说明CO32-取代了HAP晶格中的PO43-,可能是吸收了空中CO2所致。3448cm-1和1629cm-1处的吸收峰对应了H2O的伸缩振动和弯曲吸收峰,是由于在研磨压片时,压片吸收了空气中的水分引起的,因为在KBr的空白实验中该峰也存在。
从图3中可以看出,当碳量子点浓度为0.25mmol/L时可以看到颗粒均匀,分散度较好,无明显团聚现象。随着碳量子点浓度的下降,合成HAP的形貌明显变化。(a)中所制备的样品呈块状、没有规则形貌,这是由于没有碳量子点的存在将导致无定形磷酸钙盐的不可控快速生长造成的。当碳量子点浓度由0.0625mmol/L增加到1.00mmol/L,所合成的样品颗粒呈准球形,颗粒均匀(大约40nm),这一现象至少说明碳量子点的存在干预了磷酸钙盐的生长。当碳量子点浓度为0.25mmol/L,在此基础上,增加或减小碳量子点的浓度,最终产物颗粒明显发生团聚,但颗粒的尺寸基本没有发生变化。当减小碳量子点的浓度由0.0625mmol/L到0时,产物的形貌由明显的团聚到呈块状,这是由于碳量子点表面存在的羧基和羟基有限通过静电引力吸附的Ca2+达到平衡,将不再吸附带正电的Ca2+,溶液中剩余的大量Ca2+以游离态的形式存在,当向体系中加入磷源,PO43-除了与吸附在碳量子点表面的Ca2+生成无定形的磷酸钙盐沉积在其表面,同时也会与溶液中剩余的Ca2+反应无序生成无定形的磷酸钙盐,最终得到的产物除了球形的HAP纳米颗粒,还有不规则形貌的HAP。当增加碳量子点的浓度到1.00mmol/L时,这是因为加入的Ca2+不足,导致碳量子点通过共用Ca2+相互吸引,从而导致颗粒间发生团聚。由此可见,当碳量子点浓度为0.25mmol/L时,所得到的样品的最佳,颗粒均匀,无明显团聚。
图4为碳量子点浓度为0.25mmol/L时CaCl2的加入量对HAP形貌的影响。由SEM照片可以看出,当CaCl2的加入量为80mmol/L时,所得到的HAP产物颗粒均匀,大约40nm,无明显团聚现象。这是由于向体系中加入CaCl2的量较接近碳量子点表面羧基和羟基所能吸附的Ca2+的量,溶液中游离态的Ca2+较少,生成的无定形磷酸钙盐基本上都沉积在碳量子点的表面。随着溶液中CaCl2加入量的增加,产物中除了颗粒状HAP,还有少量的块状HAP出现,这是由于溶液中Ca2+过量,与加入的CaCl2的量固定,研究碳量子点浓度对其影响时,碳量子点太少原因一致。当体系中CaCl2的加入量从80mmol/L下降到40mmol/L时,可以看到颗粒发生团聚,粒径基本没有变化,这与探究碳量子点浓度对其影响时碳量子点过多引起颗粒发生团聚原因一致。
- 还没有人留言评论。精彩留言会获得点赞!
- 具有多级缓控释效果的羟基磷灰石载药微球的制备方法与流程
- 一种羟基磷灰石包覆PRB填充材料及其制备方法和地下水除铀应用方法与制造工艺
- 25‑羟基维生素D荧光免疫层析用活化荧光乳胶微球的制造方法与工艺
- 25‑羟基维生素D3荧光免疫层析用活化荧光乳胶微球的制造方法与工艺
- 壳聚糖/羟基磷灰石复合纤维的制备方法与制造工艺
- 一种羟基磷灰石‑碳纳米管复合生物陶瓷的制备方法与制造工艺
- 羟基磷灰石‑碳纳米管复合生物陶瓷的制备方法与制造工艺
- 羟基磷灰石吸附特性对水中钴离子的富集检测方法与制造工艺
- 以羟基磷灰石晶须增强可降解共聚物‑硅酸钙复合骨修复材料为原料的骨修复用制品的制造方法与工艺
- 一种胶原蛋白/羟基磷灰石复合人工骨及其制备方法与制造工艺