制备BEA型分子筛的方法及BEA型分子筛与流程
本发明属于分子筛制备领域,更具体地,涉及一种制备bea型分子筛的方法以及由该方法制备的bea型分子筛。
背景技术:
bea型分子筛是由mobil公司于1967年首先在四乙基氢氧化铵强碱体系中水热合成得到的具有三维十二元环交叉孔道体系结构的微孔高硅分子筛。bea分子筛独特的孔道结构、良好的热稳定性、水热稳定性及合适的酸度使其可作为催化材料广泛用于石油化工行业中。
考虑到bea型分子筛在实际应用中的稳定性、催化性能,要求其具有较高的si/al摩尔比(以下称“sar比”)。sar比越高,分子筛的疏水性越好,这有利于bea型分子筛在含水环境中的应用。例如,利用bea型分子筛吸附去除气体中的挥发性有机物时,低sar比的分子筛由于含有较多的酸性位点,其亲水性较强,更容易吸附气体中的水汽,从而不利于对挥发性有机物的选择性吸附,所以合成较高sar比的bea型分子筛在实际应用中具有重要的意义。
目前,直接合成具有较高sar比的bea型分子筛的方法主要有以下几种:
1)采用四乙基氢氧化铵(teaoh)等有机铵盐作为模板剂合成(专利文献1),但是该方法使用的模板剂用量较多、合成时间长、成本较高,并且所合成出来的bea型分子筛sar比一般不高于200;
2)非专利文献1、2以及专利文献2报道了采用特种模板剂合成高硅bea型分子筛的方法,但是特种模板剂的合成工艺复杂、成本较高、合成时间较长,有时甚至达到7天,合成的bea分子筛的尺寸较大,不能满足选择性吸附挥发性有机物的需求;
3)专利文献3报道了在氟离子存在的条件下,采用四乙基氢氧化铵(teaoh)作为模板剂来合成具有较高sar比的bea分子筛,但氟离子会严重腐蚀合成设备并且在后续焙烧过程中会释放出有毒的氢氟酸气体,对环境与设备的危害性非常大,而且由于合成凝胶中的水分含量很低,凝胶粘度大,无法进行搅拌混合,因而很难进行大规模生产;
4)非专利文献3报道了仅使用四乙基氢氧化铵制备全硅bea分子筛的方法,但合成时间长达10天以上,其合成效率无法进行大规模工业化生产,并且所得的分子筛不适于挥发性有机物的吸附;
5)非专利文献4提出了通过itq-1分子筛合成全硅bea分子筛的方法,该方法需要采用昂贵的金刚烷基三甲基铵盐为模板剂,虽然这种方法避免使用了氟离子、合成时间也较短,但所得的bea型分子筛形状大小不一,微孔的比表面积小于470m2/g,合成需要使用两种模板剂,并且以两种分子筛为前驱体进行合成,工序复杂,制备成本极高。
6)专利文献4、5报道了利用含有硅源与铝源的四乙基氢氧化铵溶胶,可以快速合成bea型分子筛的方法,但该方法获得的bea型分子筛的sar比为70以下,不能满足工业上作为挥发性有机物吸附剂的要求。
因此,本领域仍亟需一种能够高效率、低成本、可工业化生产高硅bea型分子筛的方法。
现有技术文献
专利文献1:us3308069
专利文献2:us5453511
专利文献3:cn104556096
专利文献4:jp5760435
专利文献5:jp5824805
非专利文献1:j.chem.soc.,chem.commun.1994,1241-1242
非专利文献2:microporousmesoporousmater.2006,89,235-245
非专利文献3:chem.commun.,2003,326–327
非专利文献4:acsappl.mater.interfaces2017,9,27273-27283
技术实现要素:
在本发明的一个方面,提供了一种制备bea型分子筛的方法,所述方法包括:
将硅源1、铝源1和模板剂1在水中混合,并且在100~180℃晶化0.5~24小时,得到晶种凝胶,所述晶种凝胶具有如下摩尔比:
si:al:q1:h2o=1:x1:y1:z1
其中0<x1<0.02,0<y1<0.6,1<z1<10,并且q1表示模板剂1;
将硅源2、铝源2和模板剂2在水中混合,并加入所述晶种凝胶,得到初始凝胶,所述初始凝胶具有如下摩尔比:
si:al:q2:h2o=1:x2:y2:z2
其中0<x2<0.02,0<y2<0.6,1<z2<10,并且q2表示模板剂2;
将所述初始凝胶在90~250℃水热合成2~72小时,分离并除去模板剂,获得bea型分子筛。
在一个实施方案中,所述晶种凝胶的添加量占所述初始凝胶的2~80重量%。在另一个实施方案中,所述晶种凝胶的添加量占所述初始凝胶的5~60重量%。在又一个实施方案中,所述晶种凝胶的添加量占所述初始凝胶的10~50重量%。
在一个实施方案中,所述方法包括将硅源1、铝源1和模板剂1在水中混合,并在0~100℃熟化2小时~30天,随后在100~180℃晶化1~12小时,得到所述晶种凝胶。在另一个实施方案中,所述方法包括将硅源1、铝源1和模板剂1在水中混合,并在10~80℃熟化3小时~10天,随后在100~180℃晶化1~12小时,得到所述晶种凝胶。在又一个实施方案中,所述方法包括将硅源1、铝源1和模板剂1在水中混合,并在25~60℃熟化5小时~4天,随后在100~180℃晶化1~12小时,得到所述晶种凝胶。
在一个实施方案中,硅源1与硅源2相同或不同,优选相同,并各自独立地选自胶态二氧化硅、无定形二氧化硅、气相二氧化硅、硅酸钠、三甲基乙氧基硅烷、正硅酸四乙酯、硅酸铝中的一种或多种。在另一个实施方案中,铝源1与铝源2相同或不同,优选相同,并各自独立地选自硫酸铝、硝酸铝、铝酸钠、氧化铝、氢氧化铝、勃姆石、氯化铝、硅酸铝、金属铝中的一种或多种。在又一个实施方案中,模板剂1与模板剂2相同或不同,优选相同,并各自独立地选自四甲基氢氧化铵、四乙基氢氧化铵、四丙基氢氧化铵、四丁基氢氧化铵、二甲基二乙基氢氧化铵中的一种或多种。
在一个实施方案中,所述初始凝胶在经历水热合成之前的含水量为10~80重量%。在另一个实施方案中,所述初始凝胶在经历水热合成之前的含水量为30~70重量%。在一个实施方案中,所述初始凝胶在经历水热合成之前在0~100℃熟化2小时~30天。在另一个实施方案中,所述初始凝胶在经历水热合成之前在10~80℃熟化3小时~10天。在又一个实施方案中,所述初始凝胶在经历水热合成之前在25~60℃熟化5小时~4天。
在一个实施方案中,所述水热合成在100~200℃进行4~72小时。在另一个实施方案中,所述水热合成在120~180℃进行12~72小时。在一个实施方案中,通过煅烧方法或有机溶剂提取方法来去除模板剂。在另一个实施方案中,通过在空气或含氧的惰性气氛下、于300~1000℃、优选400~900℃、更优选450~850℃、还更优选500~800℃煅烧,从而除去模板剂。
在本发明的另一方面,提供了一种bea型分子筛,其中si/al比为50以上,总比表面积为550m2/g以上,微孔比表面积为480m2/g以上,粒径为200nm以下。在一个实施方案中,提供了一种由本文所述的方法制备的bea型分子筛,其中si/al比为50以上,总比表面积为550m2/g以上,微孔比表面积为480m2/g以上,粒径为200nm以下。在另一个实施方案中,在固体29si核磁谱图中,所述bea型分子筛的-106~-95ppm峰的面积占-120~-95ppm的峰面积的22%以下。
在本发明的又一方面,提供了一种气体处理系统,其包括根据本文所述的方法制备得到的bea型分子筛或者本文所述的bea型分子筛。
在本发明的进一方面,提供了根据本文所述的方法制备得到的bea型分子筛或者本文所述的bea型分子筛在选择性吸附挥发性有机物中的用途。
通过本文所述的方法,可以在使用常规原料且不使用含氟化合物或特殊模板剂的情况下,在较短时间内合成具有高sar比的bea型分子筛,并且所得的bea型分子筛具有均一的相貌、高结晶度、低杂质含量、高微孔比表面积等特点,有利于在含有水汽的条件下,选择性大量吸附工业废气中的挥发性有机物。
附图说明
本文提供了附图,以使本领域技术人员更好地理解本文所例示的实施方式,但是这些附图不旨在限定本发明的范围。
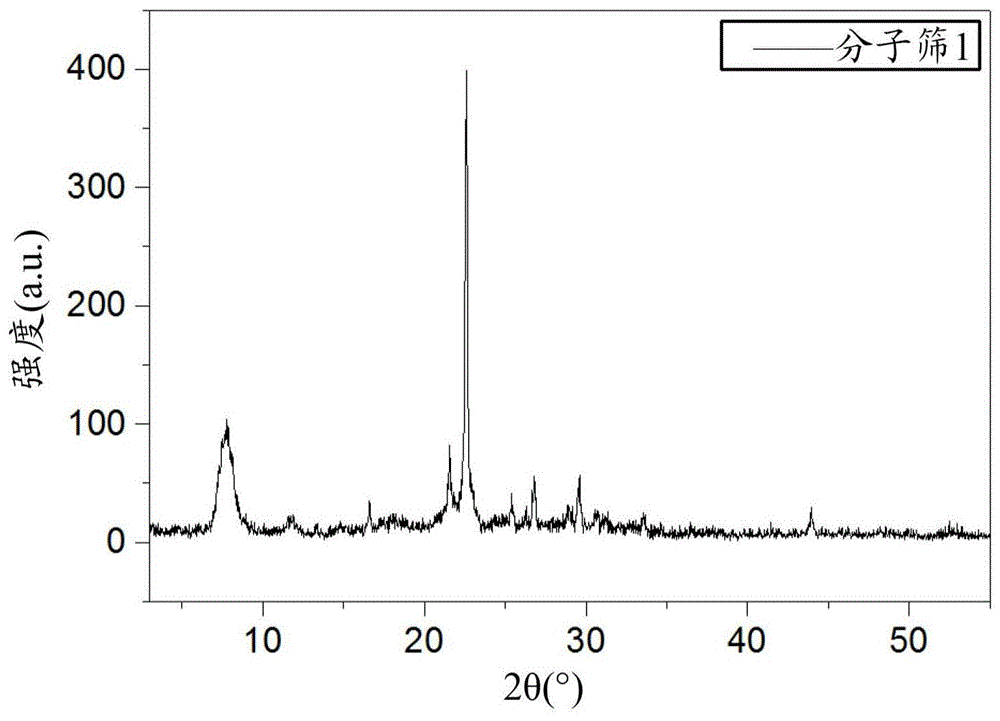
图1是根据本发明的方法制备的分子筛1的xrd谱图;
图2是根据本发明的方法制备的分子筛2的xrd谱图;
图3是根据本发明的方法制备的分子筛2的固体29si核磁谱图;
图4是根据本发明的方法制备的分子筛2的sem谱图;
图5是根据本发明的方法制备的分子筛4的固体核磁29si谱图;
图6是根据本发明的方法制备的分子筛8的固体核磁29si谱图;
图7是根据比较例的方法制备的分子筛9的xrd谱图;
图8是根据比较例的方法制备的分子筛10的xrd谱图;
图9是根据比较例的方法制备的分子筛11的xrd谱图。
具体实施方式
在下文中,将通过具体实施方案来进一步阐述本发明的内容。然而,所列举的具体实施方案仅出于例示目的,而并不旨在限制本发明的范围。本领域技术人员会认识到,以下任一实施方案中所给出的具体技术特征可以用于其他实施方案,只要其不背离本发明的主旨即可。
在一个实施方案中,制备bea型分子筛的方法包括:
将硅源1、铝源1和模板剂1在水中混合,并且在100~180℃晶化0.5~24小时,得到晶种凝胶,所述晶种凝胶具有如下摩尔比:
si:al:q1:h2o=1:x1:y1:z1
其中0<x1<0.02,0<y1<0.6,1<z1<10,并且q1表示模板剂1;
将硅源2、铝源2和模板剂2在水中混合,并加入所述晶种凝胶,得到初始凝胶,所述初始凝胶具有如下摩尔比:
si:al:q2:h2o=1:x2:y2:z2
其中0<x2<0.02,0<y2<0.6,1<z2<10,并且q2表示模板剂2;
将所述初始凝胶在90~250℃水热合成2~72小时,分离并除去模板剂,获得bea型分子筛。在另一个实施方案中,将硅源2、铝源2和模板剂2在水中混合,得到与晶种凝胶中的si:al:q1比例相同的混合凝胶,之后再加入所述晶种凝胶。采用这样的方法,可以更好地控制和调整最终获得的分子筛成分和结构。
在一个实施方案中,所述混合凝胶可以在0~100℃熟化2小时~30天。在另一个实施方案中,所述混合凝胶在10~80℃熟化3小时~10天。在又一个实施方案中,所述混合凝胶在25~60℃熟化5小时~4天。当然,混合凝胶可以在制备后无需熟化,也可以在上述范围内进行熟化。
在上述方法中,硅源1、铝源1和模板剂1的混合顺序没有特别的限制,只要能够均匀混合即可。例如,可以将硅源添加到含模板剂的水溶液中,然后再加入铝源,待混合均匀后,将混合物置于密闭容器中,在100~180℃反应0.5~24小时,制备得到透明水性凝胶,其可用作后续步骤的晶种(即,晶种凝胶,有时也可称为水性凝胶)。进一步地,在将硅源、铝源和模板剂混合之后加入晶种凝胶,待混合均匀后得到初始凝胶,该初始凝胶可被置于密闭容器中,在90~250℃水热合成2~72小时。随后,将含有结晶分子筛的母液进行分离、清洗、干燥,并除去分子筛中的模板剂,即可得到具有高sar比的bea型分子筛。
在一个实施方案中,硅源1与硅源2可以彼此相同或不同,优选相同。在本发明中,硅源不受特别的限制,只要能够与其它成分均匀混合即可。在另一个实施方案中,硅源可以选自胶态二氧化硅、无定形二氧化硅、气相二氧化硅、硅酸钠、三甲基乙氧基硅烷、正硅酸四乙酯、硅酸铝中的一种或多种。然而,需要说明,如果硅源是液体,只要其像二氧化硅溶胶那样已经形成5~60重量%二氧化硅的水分散液,就可以使用该液体。当采用这种液体时,优选采用以硅原子浓度记为5重量%以上、特别是10重量%以上且80重量%以下、特别是50重量%以下的水分散液。
在一个实施方案中,铝源1与铝源2可以彼此相同或不同,优选相同。在本发明中,铝源不受特别的限制,只要能够与其它成分均匀混合即可。在另一个实施方案中,铝源可以选自硫酸铝、硝酸铝、铝酸钠、氧化铝、氢氧化铝、勃姆石、氯化铝、硅酸铝、金属铝中的一种或多种。
在一个实施方案中,模板剂1与模板剂2可以彼此相同或不同,优选相同。在本发明中,模板剂不受特别的限制,只要其能够有效地形成bea型分子筛即可。在另一个实施方案中,模板剂是季铵盐类化合物,优选地,模板剂可以选自四甲基氢氧化铵、四乙基氢氧化铵、四丙基氢氧化铵、四丁基氢氧化铵、二甲基二乙基氢氧化铵中的一种或多种。
在一个实施方案中,除了硅源、铝源和模板剂之外,还可以采用其它原料,由此制得的bea型分子筛除了含有硅铝作为分子筛的骨架原子之外,还可以包含其它元素原子。在另一个实施方案中,所述其它元素选自碱金属、碱土金属、铜、钛、锆、钒、铬、锰、铁、钴、锌、镓、锗、砷、锡和硼中的一种或多种。优选地,所述其它元素是碱金属或碱土金属。在又一个实施方案中,所述其它元素可以在制备晶种凝胶时被引入,或在制备初始凝胶时被引入。
在一个实施方案中,所述方法包括将硅源1、铝源1和模板剂1在水中混合,并在0~100℃熟化2小时~30天,随后在100~180℃晶化1~12小时,得到所述晶种凝胶。在另一个实施方案中,所述方法包括将硅源1、铝源1和模板剂1在水中混合,并在10~80℃熟化3小时~10天,随后在100~180℃晶化1~12小时,得到所述晶种凝胶。在又一个实施方案中,所述方法包括将硅源1、铝源1和模板剂1在水中混合,并在25~60℃熟化5小时~4天,随后在100~180℃晶化1~12小时,得到所述晶种凝胶。
当然,上述的熟化过程并非是必须的,即,可以先进行熟化,也可以不经历熟化而直接进行晶化反应。在熟化过程中,熟化温度可以是恒定的,也可以是跃变或渐变的。
在一个实施方案中,所述方法包括将硅源1、铝源1和模板剂1在水中混合,并在100~180℃晶化1~12小时,得到所述晶种凝胶。在另一个实施方案中,所述方法包括将硅源1、铝源1和模板剂1在水中混合,并在120~160℃晶化3~10小时,得到所述晶种凝胶。晶化温度过低和/或时间过短,不能导致硅源与铝源的充分溶解,而晶化温度过高和/或时间过长,不仅会导致制造成本上升,还会导致模板剂分解,以及沉淀物的生成,不能得到透明水性凝胶。
对于晶种凝胶的含水量,鉴于硅源和铝源的均一溶解与分散的容易程度、制造成本方面考虑,含水量为10重量%以上,优选为20重量%以上,并且为80重量%以下、特别优选为70重量%以下。
在一个实施方案中,所述晶种凝胶的添加量占所述初始凝胶的2~80重量%。在另一个实施方案中,所述晶种凝胶的添加量占所述初始凝胶的5~60重量%。在又一个实施方案中,所述晶种凝胶的添加量占所述初始凝胶的10~50重量%。在一个实施方案中,所述初始凝胶在经历水热合成之前的含水量为10~80重量%。在另一个实施方案中,所述初始凝胶在经历水热合成之前的含水量为30~70重量%。
在一个实施方案中,所述初始凝胶在经历水热合成之前在0~100℃熟化2小时~30天。在另一个实施方案中,所述初始凝胶在经历水热合成之前在10~80℃熟化3小时~10天。在又一个实施方案中,所述初始凝胶在经历水热合成之前在25~60℃熟化5小时~4天。当然,初始凝胶可以在制备后立即进行水热合成反应而无需熟化,也可以在上述范围内进行熟化。
在一个实施方案中,所述水热合成在90~250℃进行2~72小时。在又一个实施方案中,所述水热合成在100~200℃进行4~72小时,如12、18、24、36、48、72小时。在另一个实施方案中,所述水热合成在120~180℃进行12~72小时,如12、18、24、36、48、72小时。在其它实施方案中,所述水热合成在150~160℃进行12~72小时,如12、18、24、36、48、72小时。水热合成的反应温度可以是恒定的,也可以是跃变或渐变的。分子筛的该水热合成温度过高和/或时间过长,不仅会导致模板剂分解,还会导致生成杂项,产物中bea分子筛的纯度下降。分子筛的该水热合成温度过低和/或时间过短,会导致bea分子筛的产率降低,制造成本增加。
在本发明中,将水热合成反应后的产物分离的方法不受特别的限制,通常可以采用过滤、倾析、离心、膜分离或直接干燥等。为了将制备时使用的模板剂(如季铵盐)去除,从水热合成反应液分离回收的含有模板的bea型分子筛可以根据需要在水洗、干燥后进行煅烧等后处理。将本发明的bea型分子筛用于催化剂(也包括催化剂载体)、吸附材料等用途时,优选在将模板剂及制备过程中引入的钠等碱金属或碱土金属元素去除后再使用。去除模板和/或碱金属或碱土金属的后处理可以采用使用酸性溶液、包含模板分解成分的化学溶液的液相处理,使用树脂等的离子交换处理,热分解处理,也可以组合使用这些处理。
在一个实施方案中,通过煅烧方法或有机溶剂提取方法来去除模板剂。在另一个实施方案中,通过在空气或含氧的惰性气氛下、于300~1000℃、优选400~900℃、更优选450~850℃、还更优选500~800℃煅烧,从而除去模板剂。在本发明中,惰性气氛优选采用氮气。
在获得bea型分子筛之后,也可以利用分子筛的离子交换能力将碱金属部分变换成h型、nh4型而使用,其方法可以采用公知的技术。可以通过利用nh4no3、nh4cl等铵盐或者盐酸等酸,在室温至100℃的温度下进行上述处理,随后进行水洗、烘干等处理。
本发明的bea型分子筛是指国际分子筛协会(internationalzeoliteassociation,以下简称iza)规定的bea型分子筛。分子筛通常为由骨架原子四面体(如sio4四面体、alo4四面体或者po4四面体,并且通常将氧元素之外的元素原子称为非氧原子或t原子)的各个顶点的氧原子通过共用联结而成的规则网目状结构。网目结构的基本单元之一是n个to4四面体连接而成的环状。是否为本发明的分子筛可以通过如下分析来判断:
首先,对试样进行x射线衍射(xrd)测定,如至少检测到表1所示的晶面间距
<表1>
之后,通过将试样在酸性溶液中溶解后进行元素组成分析,测定骨架元素硅和铝的含量,从而确认其sar比。也可以通过固体29si核磁谱图,计算出分子筛骨架中硅和铝的摩尔比,从而确认sar比。
之后,对试样进行77k氮气吸附等温线测定,求出其bet比表面积以及微孔比表面积。最后通过扫描电子显微镜(sem),确定试样的粒径。
如果xrd、bet、固体29si核磁谱图以及扫描电子显微镜的结果均满足本发明的规定,则能够判断该分子筛为本发明的分子筛。
在一个实施方案中,提供了一种bea型分子筛,其中si/al比为50以上,总比表面积为550m2/g以上,微孔比表面积为480m2/g以上,粒径为200nm以下。在另一个实施方案中,提供了一种由本文所述的方法制备的bea型分子筛,其中si/al比为50以上,总比表面积为550m2/g以上,微孔比表面积为480m2/g以上,粒径为200nm以下。在又一个实施方案中,在固体29si核磁谱图中,所述bea型分子筛的-106~-95ppm峰的面积占-120~-95ppm的峰面积的22%以下。
在一个实施方案中,所述bea型分子筛的si/al比为80以上,优选90以上,更优选100以上。在另一个实施方案中,所述bea型分子筛的总比表面积为700m2/g以上。在另一个实施方案中,所述bea型分子筛的微孔比表面积为600m2/g以上。
在一个实施方案中,除了硅、铝、氧之外,bea型分子筛还包含其它元素作为分子筛的骨架原子。在另一个实施方案中,所述其它元素选自碱金属、碱土金属、铜、钛、锆、钒、铬、锰、铁、钴、锌、镓、锗、砷、锡和硼中的一种或多种。优选地,所述其它元素是碱金属或碱土金属。
在一个实施方案中,提供了一种气体处理系统,其包括根据本文所述的方法制备得到的bea型分子筛或者本文所述的bea型分子筛。在一个实施方案中,所述气体处理设备是挥发性有机物吸附设备。
举例而言,可以将本文的bea型分子筛与有机和/或无机粘合剂混合配制成浆料,浸渍涂敷到蜂窝状转轮上。也可以将本文的bea分子筛、有机和/或无机粘合剂、有机和/或无机纤维混合后抄纸,再经过定型制备出瓦楞纸,最后制备出含有bea分子筛的蜂窝状转轮。另外,还可以将bea分子筛造粒成型后,装入吸附塔中,用于特定物质(如发挥性有机物)的吸附。
在一个实施方案中,所述气体处理设备是挥发性有机气体处理设备。含有本文的bea型分子筛的挥发性有机气体处理设备可以对含有低浓度挥发性有机物的气体进行吸附浓缩,然后再通过升温脱附,减压脱附等手段,将高浓度的挥发性有机物进行蓄热燃烧,催化燃烧等无害化处理,从而有效去除气体中的低浓度挥发性有机气体。
在一个实施方案中,提供了根据本文所述的方法制备得到的bea型分子筛或者本文所述的bea型分子筛在选择性吸附挥发性有机物中的用途。
实施例
在下文,通过具体的实施例对本发明进行更进一步的说明,但本发明并不仅限于以下实施例。在以下实施例中,所用的化学试剂如未特别说明,均为化学纯。
粉末xrd的测定
使用日本理学制造的x射线粉末衍射仪测定装置(ulitmaiv)进行测定。
元素组分的测定
使用赛默飞世尔科技(中国)有限公司制造的icp全谱直读发射光谱测定装置(irisadvantage)进行测定。
bet比表面积的测定
使用贝士德仪器科技(北京)有限公司制造的高性能比表面及微孔径分析仪(3h-2000)进行测定。采用bet法测得总比表面积,采用t-plot法测得微孔比表面积,总比表面积减去微孔比表面积即为外比表面积。一般来说,样品的微孔比表面积越高,说明样品的结晶性越好。
固体核磁的测定
使用varian400宽腔固体谱仪,29simas转速8000/秒,弛豫延迟4秒,每次样品的采样次数不固定,29si的共振频率79mhz,采用π/2激发脉冲。27almas转速8000/秒,弛豫延迟1秒,每个样品的采样次数不固定,27al的共振频率104mhz,采用π/12激发脉冲。
表面形貌及粒径的测定
使用由zeiss制造的扫描电子显微镜(merlincompact)观测。
本发明中的粒径通过随机拍摄试样的sem图,并从图中随机选择50个颗粒进行粒径统计,求出其平均尺寸。
实施例1
将28.06g四乙基氢氧化铵(teaoh含量:30wt%)溶液和25gjn-40硅溶胶溶液(sio2含量:40wt%,青岛海洋公司制)配成混合溶液,搅拌10分钟后将0.0632gal(oh)3加入到上述混合溶液中,室温搅拌并熟化12小时。随后,将所得的产物置于烘箱中,并按照晶种凝胶sio2:0.002al2o3:0.4teaoh:6.5h2o的摩尔比蒸发水分,将产物装入带聚四氟乙烯内衬的不锈钢晶化釜中,于150℃晶化1小时,得到晶种凝胶。
按上述相同方法配制摩尔比sio2:0.002al2o3:0.4teaoh:4.5h2o的凝胶,将该凝胶在室温搅拌熟化24小时,然后置于烘箱中按照上述摩尔比蒸发水分,获得具有上述摩尔比的凝胶,加入上述晶种凝胶,搅拌30分钟,获得初始凝胶。将初始凝胶装入带聚四氟乙烯内衬的不锈钢晶化釜中,于160℃水热合成72小时。反应结束后,冷却产物,过滤回收晶体。将回收的晶体在100℃下干燥12小时,得到bea型分子筛1,对其进行xrd测定,结果显示出如表2所示的位置和相对强度。该分子筛1的xrd谱图示于图1中。
表2
进而,将bea型分子筛1在600℃的空气气流下煅烧6小时,将模板剂去除,得到bea型分子筛2。对该分子筛2进行xrd测定,结果显示出如表3所示的位置和相对强度。该分子筛2的xrd谱图示于图2中。
表3
分子筛2的元素组分测定的结果显示,sar比为336。
分子筛2测定的固体核磁29si谱图示于图3中,其中29si谱在-106~-95ppm间的峰面积占-120~-95ppm的峰面积的8.3%。
bet测定结果:总比表面积为720m2/g,外比表面积为65m2/g,微孔比表面积为655m2/g。分子筛2的sem图示于图4中。
该方法合成的bea型分子筛,其微孔比表面积高,相较于以往不加透明凝胶合成的bea型分子筛,结晶性更好。
实施例2
将28.06g四乙基氢氧化铵(teaoh含量:30wt%)溶液和25gjn-40硅溶胶溶液(sio2含量:40wt%,青岛海洋公司制)配成混合溶液,搅拌10分钟后将0.0632gal(oh)3加入到上述混合溶液中,室温搅拌并熟化12小时。随后,将所得的产物置于烘箱中,并按照晶种凝胶sio2:0.002al2o3:0.4teaoh:6.5h2o的摩尔比蒸发水分,将产物装入带聚四氟乙烯内衬的不锈钢晶化釜中,于150℃晶化3小时,得到晶种凝胶。
按上述相同方法配制摩尔比sio2:0.002al2o3:0.4teaoh:4.4h2o的凝胶,将该凝胶在室温搅拌熟化24小时,然后置于烘箱中按照上述摩尔比蒸发水分,获得具有上述摩尔比的凝胶,加入上述晶种凝胶,搅拌30分钟,获得初始凝胶。将初始凝胶装入带聚四氟乙烯内衬的不锈钢晶化釜中,于160℃水热合成72小时。反应结束后,冷却产物,过滤回收晶体。将回收的晶体在100℃下干燥12小时,得到bea型分子筛3,对其进行xrd测定,结果显示出如表2所示的位置和相对强度。该分子筛3的xrd谱图与分子筛1的xrd谱图一致。
进而,将bea型分子筛3在600℃的空气气流下煅烧6小时,将模板剂去除,得到bea型分子筛4。对该分子筛4进行xrd测定,结果显示出如表3所示的位置和相对强度。该分子筛4的xrd谱图与分子筛2的xrd谱图一致。
分子筛4的元素组分测定的结果显示,sar比为341。
分子筛4测定的固体核磁29si谱图示于图5中,其中29si谱在-106~-95ppm间的峰面积占-120~-95ppm的峰面积的9.1%。
bet测定结果:总比表面积为740m2/g,外比表面积为86m2/g,微孔比表面积为654m2/g。分子筛4的sem图与分子筛2的sem图形貌一致。
该分子筛同样相较于以往不加透明凝胶合成的bea型分子筛微孔比表面积更高,结晶性更好。
实施例3
将28.06g四乙基氢氧化铵(teaoh含量:30wt%)溶液和25gjn-40硅溶胶溶液(sio2含量:40wt%,青岛海洋公司制)配成混合溶液,搅拌10分钟后将0.0632gal(oh)3加入到上述混合溶液中,室温搅拌并熟化12小时。随后,将所得的产物置于烘箱中,并按照晶种凝胶sio2:0.002al2o3:0.4teaoh:6.5h2o的摩尔比蒸发水分,将产物装入带聚四氟乙烯内衬的不锈钢晶化釜中,于150℃晶化0.5小时,得到晶种凝胶。
按上述相同方法配制摩尔比sio2:0.002al2o3:0.4teaoh:4.4h2o的凝胶,将该凝胶在室温搅拌熟化24小时,然后置于烘箱中按照上述摩尔比蒸发水分,获得具有上述摩尔比的凝胶,加入上述晶种凝胶,搅拌30分钟,获得初始凝胶。将初始凝胶装入带聚四氟乙烯内衬的不锈钢晶化釜中,于160℃水热合成72小时。反应结束后,冷却产物,过滤回收晶体。将回收的晶体在100℃下干燥12小时,得到bea型分子筛5,对其进行xrd测定,结果显示出如表2所示的位置和相对强度。该分子筛5的xrd谱图与分子筛1的xrd谱图一致。
进而,将bea型分子筛5在600℃的空气气流下煅烧6小时,将模板剂去除,得到bea型分子筛6。对该分子筛6进行xrd测定,结果显示出表3所示的位置和相对强度。该分子筛6的xrd谱图与分子筛2的xrd谱图一致。
bet测定结果为:总比表面积为720m2/g,外比表面积为65m2/g,微孔比表面积655m2/g。
该分子筛同样相较于以往不加透明凝胶合成的bea型分子筛微孔比表面积更高,结晶性更好。
<实施例4>
将28.06g四乙基氢氧化铵(teaoh含量:30wt%)溶液和25gjn-40硅溶胶溶液(sio2含量:40wt%,青岛海洋公司制)配成混合溶液,搅拌10分钟后将0.3160gal(oh)3加入到上述混合溶液中,室温搅拌并熟化12小时。随后,将所得的产物置于烘箱中,并按照晶种凝胶sio2:0.01al2o3:0.4teaoh:6.5h2o的摩尔比蒸发水分,将产物装入带聚四氟乙烯内衬的不锈钢晶化釜中,于150℃晶化5小时,得到晶种溶胶。
按上述相同方法配制摩尔比sio2:0.01al2o3:0.4teaoh:4.5h2o的凝胶,将该凝胶在室温搅拌熟化24小时,然后置于烘箱中按照上述摩尔比蒸发水分,获得具有上述摩尔比的凝胶,加入上述晶种凝胶,搅拌30分钟,获得初始凝胶。将初始凝胶装入带聚四氟乙烯内衬的不锈钢晶化釜中,于160℃水热合成72小时。反应结束后,冷却产物,过滤回收晶体。将回收的晶体在100℃下干燥12小时,得到bea型分子筛7,对其进行xrd测定,结果显示出如表2所示的位置和相对强度。
进而,将bea型分子筛7在600℃的空气气流下煅烧6小时,将模板剂去除,得到bea型分子筛8。对该分子筛8进行xrd测定,结果显示出表3所示的位置和相对强度。该分子筛8的xrd谱图与分子筛2的xrd谱图一致。
分子筛8的元素组分测定的结果显示,sar比为99.9。
分子筛8测定的固体核磁29si谱图示于图6中,其中29si谱在-106.1~-94.9ppm间的峰面积占-119.8~-94.9ppm的峰面积的22%。
bet测定结果:总比表面积为733m2/g,外比表面积为103m2/g,微孔比表面积630m2/g。
该分子筛同样相较于以往不加透明凝胶合成的bea型分子筛微孔比表面积更高,结晶性更好。
实施例5
本实施例与上述实施例1大致相同,但采用的水热合成反应时间为12小时,获得纯相的bea型分子筛。元素组分测定的结果:sar比为348.3。
实施例6
本实施例与实施例1大致相同,但采用的水热合成反应时间为36小时,获得纯相的bea型分子筛。元素组分测定的结果:sar比为207.3。
实施例7
本实施例步骤与实施例1大致相同,但采用的反应釜为带搅拌的大反应釜,加入的溶胶的总量按比例扩大8倍,水热合成反应时间为36小时,获得纯相的bea型分子筛。元素组分测定的结果:sar比为370。
比较例1
在实施例1所述的初始凝胶制备过程中,不添加晶种凝胶,其余按照实施例1相同的方法进行水热合成,得到分子筛9。其xrd如图7所示,未能获得纯相的bea型分子筛,所制备的样品中,除了含有bea型分子筛之外,还含有mfi杂相。
比较例2
在实施例1所述的初始凝胶制备过程中,不添加晶种凝胶,而是添加bea型分子筛晶种(日本东曹公司,bea(980hoa),sar500,球状,粒径500-1000nm),其余按照实施例1相同的方法进行水热合成,得到分子筛10。其xrd如图8所示,未能获得纯相的bea型分子筛,所制备的样品中,除了含有bea型分子筛之外,还含有mtw杂相。
比较例3
在实施例1所述的初始凝胶制备过程中,不添加晶种凝胶,而是添加bea型分子筛晶种(南开催化剂厂,sar30),其余按照实施例1相同的方法进行水热合成,得到样品分子筛11。其xrd如图9所示,未能获得纯相的bea型分子筛,所制备的样品为zsm-5(mfi)。
尽管上文参考特定的实施例来描述本发明的具体实施方式,但是应当理解,本领域技术人员会对其作出多种调整和改变,只要其不违背本发明的范围和主旨即可。
- 还没有人留言评论。精彩留言会获得点赞!