(甲基)丙烯酰胺化合物的前体的制作方法与工艺
(甲基)丙烯酰胺化合物的前体发明领域本发明涉及(甲基)丙烯酰胺化合物的前体化合物。发明背景(甲基)丙烯酰胺是表现出高反应性的可聚合化合物。这些化合物广泛地用作工业应用例如涂料、油漆、印刷油墨、粘接剂和抗蚀剂材料中的不同种类的合成树脂的原材料或者交联剂。例如,含有(甲基)丙烯酰胺作为可自由基聚合化合物的油墨组合物描述在JP-A-2005-307198中(“JP-A”表示未审查公开的日本专利申请)。(甲基)丙烯酰胺通常是通过胺化合物与(甲基)丙烯酸酯反应来获得的。但是,该方法导致了低的产率,因为烷基胺化合物进一步与由此获得的酰胺化的化合物的(甲基)丙烯酸类基团反应,或者这是进一步的酰胺化。此外,已知的是这样的生产(甲基)丙烯酰胺的方法:将烷基胺与烷基酸烷基反应来将它们转化成氨基酰胺(酰胺加成物),并且热分解产物或者调整单烷基胺的浓度(例如JP-A-4-208258和美国专利No.2683741)。但是,当多个(甲基)丙烯酰胺基团包括在一个分子中时,这些方法仍然具有抑制副产物的问题。在所述的情形下,期望的是新的前体(合成中间体),其用于有效地生产具有多个(甲基)丙烯酰胺基团的化合物,并且其用于不同种类的合成树脂特别是油墨组合物的原材料或者交联剂。
技术实现要素:
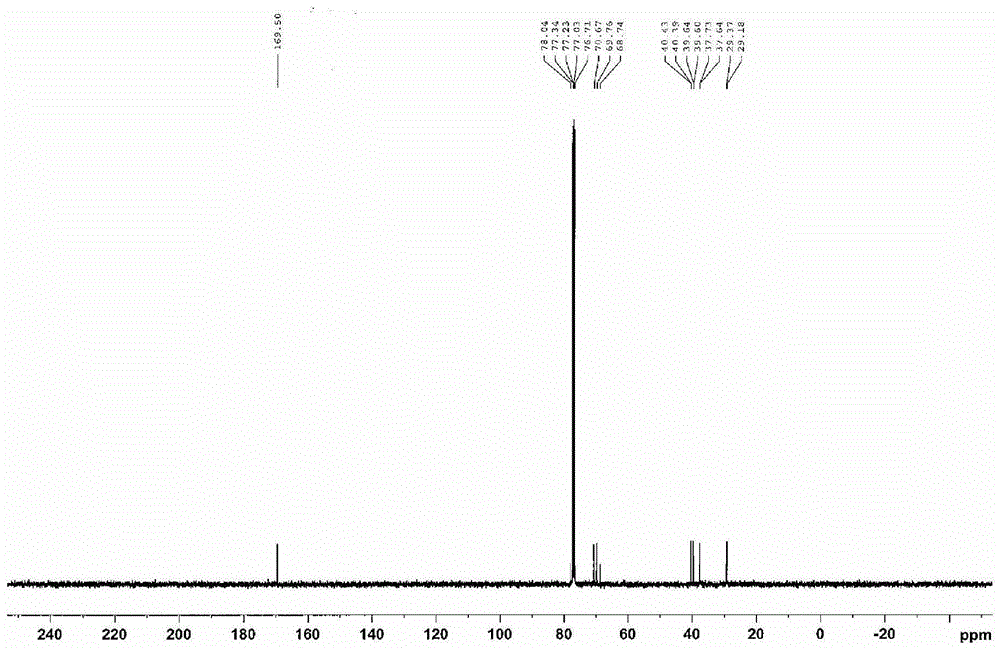
本发明在于式(1)所示的化合物:式(1)其中在式(1)中,Z代表通过从多元醇的羟基中除去n个氢原子而获得的残基;n代表3-6的整数;R1代表氢原子或者甲基;R2代表具有1-8个碳原子的亚烷基;X1代表卤素原子;和多个R1、R2和X1可以彼此相同或者不同。本发明的其他和进一步的目标、特征和优点将从下面的说明,适当的参考附图而变得更显而易见。附图说明图1是在后述的实施例1中合成的示例性化合物(1)的1H-NMR光谱图。图2是在实施例1中合成的示例性化合物(1)的13C-NMR光谱图。图3是在实施例1中合成的示例性化合物(1)的IR光谱图。图4是在实施例1中合成的示例性化合物(1)的MS光谱图。图5是在后述的实施例2中合成的示例性化合物(2)的1H-NMR光谱图。图6是在实施例2中合成的示例性化合物(2)的13C-NMR光谱图。图7是在实施例2中合成的示例性化合物(2)的IR光谱图。图8是在实施例2中合成的示例性化合物(2)的MS光谱图。具体实施方式本发明的发明人已发现化合物可以用作(甲基)丙烯酰胺化合物的合成前体,并且与常规方法相比,根据使用该前体的合成方法可以实现较高的产率。基于这一发现完成了本发明。根据本发明,提供了下面的手段:<1>式(1)所示的化合物:式(1)其中在式(1)中,Z代表通过从多元醇的羟基中除去n个氢原子而获得的残基;n代表3-6的整数;R1代表氢原子或者甲基;R2代表具有1-8个碳原子的亚烷基;X1代表卤素原子;和多个R1、R2和X1可以彼此相同或者不同。<2>根据上述项目<1>的化合物,其中该化合物是式(A)所示的(甲基)丙烯酰胺化合物的合成中间体:式(A)其中在式(A)中,Z代表通过从多元醇的羟基中除去n个氢原子而获得的残基;n代表3-6的整数;R1代表氢原子或者甲基;R2代表具有1-8个碳原子的亚烷基;和多个R1和R2可以彼此相同或者不同。在说明书中,术语“(甲基)丙烯酰胺”表示丙烯酰胺和/或甲基丙烯酰胺。此外,在说明书中,“-”表示包括将在它之前和之后的数值作为最小值和最大值的一个范围。[前体化合物]本发明的化合物是用下式(1)表示的,并且可以用作合成可聚合化合物例如(甲基)丙烯酰胺化合物的前体(合成中间体)。式(1)在式(1)中,Z代表通过从多元醇的羟基中除去n个氢原子而获得的残基。该多元醇优选是具有3-6个羟基的多羟基醇,更优选具有3-5个羟基的多羟基醇,和仍然更优选具有3-4个羟基的多羟基醇。该多元醇优选具有3-12个碳原子,更优选3-10个碳原子,和特别优选3-6个碳原子。此外,该多元醇可以是通过多羟基醇化合物的两个或者更多个分子的分子间缩合(脱水)而形成的多羟基醇缩合化合物。多元醇具体的例子包括甘油,三羟甲基乙烷,三羟甲基丙烷,二(三羟甲基)丙烷,木糖醇,山梨糖醇,赤藓醇,季戊四醇,二季戊四醇,甘露糖醇和三(2-羟乙基)异氰脲酸酯。在多元醇中,甘油、赤藓醇或者季戊四醇是优选的。在式(1)中,n代表3-6的整数。n优选是3-5的整数,和更优选3-4的整数。在式(1)中,R1代表氢原子或者甲基。R1优选是氢原子。多个R1可以彼此相同或者不同,并且优选相同。在式(1)中,R2代表具有1-8个碳原子的亚烷基。该亚烷基可以是直链或者支链。R2优选是具有2-5个碳原子的亚烷基,和更优选具有3-4个碳原子的亚烷基。多个R2可以彼此相同或者不同,并且优选相同。在式(1)中,X1代表卤素原子。X1优选是碘原子、溴原子或者氯原子,和特别优选氯原子。多个X1可以彼此相同或者不同,并且优选相同。式(1)所示的化合物特别优选是下式(2)或者式(3)所示的化合物。式(2)式(3)在式(2)和式(3)中,R1和X1每个具有与式(1)中的R1和X1相同的含义,并且R1和X1优选的范围也与式(1)中的R1和X1的相同。多个R1和X1可以彼此相同或者不同。下文中,显示了本发明的式(1)所示化合物的具体例子,但是本发明不限于此。[前体化合物的合成方法]合成式(I)所示化合物的方法的具体例子包括下面的通用合成方法,其使用胺化合物作为起始材料。·合成方法1一种在碱存在下将胺化合物与酰基卤化合物进行反应的方法。·合成方法2一种在碱存在下将胺化合物与羧酸化合物和缩合剂进行反应的方法。·合成方法3一种通过加热胺化合物和酯化合物根据酯-酰胺交换反应来合成前体化合物的方法。这些反应可以根据ShinJikkenKagakuKoza(NewExperimentalChemistryCourse)14,SynthesisandReactionofOrganicCompounds(V),11.6:ProtectionofAminoGroups,第2555-2569页中所述的方法来进行。用于上述合成方法中的胺化合物的例子包括下面的化合物。这些胺化合物可以使用市售品,或者可以根据通常已知的反应(例如胺的取代反应;硝基、叠氮化物或者腈的还原反应;酰胺、亚胺或者异氰酸酯的水解反应)使用可以是胺化合物的合成前体的化合物来合成。具体地,市售多元醇可以通过将作为该多元醇的羟基的-OH用(甲基)丙烯腈转化成-OCH2CH(R1)CN,然后还原所获得的腈基来形成-OCH2CH(R1)CH2NH2来合成的。这里,R1代表氢原子或者甲基。具体的例子在下文中给出。[使用前体化合物合成(甲基)丙烯酰胺化合物的方法]本发明的式(I)所示的化合物可以作为前体,用于合成式(A)所示的(甲基)丙烯酰胺化合物。在式(A)中,Z代表通过从多元醇的羟基中除去n个氢原子而获得的残基;n代表3-6的整数;R1代表氢原子或者甲基;R2代表具有1-8个碳原子的亚烷基;和多个R1和R2可以彼此相同或者不同。在使用本发明的式(1)所示的化合物(下文中,该化合物称作本发明的前体化合物)合成(甲基)丙烯酰胺化合物中,例如该(甲基)丙烯酰胺化合物可以如下来获得:将碱(有机碱和/或无机碱)作用于本发明的前体化合物上,并且根据前体的X1和键合到与R1键合的碳原子上的氢原子之间的消去反应在末端形成碳-碳双键。在下面的合成方案1中,表示了用于由胺化合物来合成本发明的前体化合物[在下面的方案中用式(2)所示的化合物],和进一步由该前体来合成(甲基)丙烯酰胺化合物1的方法具体的例子。另外,R1和X1在下面的合成方案中具有与上述式(2)中的R1和X1相同的含义。合成方案1根据上述合成方案,首先,将甘油作为起始材料(A)用于使得甘油的羟基与丙烯腈或者甲基丙烯腈反应,和因此获得了作为中间体(B)的(甲基)丙烯腈加成物。接着,将所获得的中间体(B)与氢在催化剂存在下反应,和因此根据氢化反应获得了作为中间体(C)的胺形式。该所获得的中间体(C)进一步与3-氯丙酰基氯或者3-氯-2-甲基丙酰基氯反应来进行酰胺化,和因此能够获得作为本发明前体化合物的式(2)所示的化合物。作为酰胺化试剂,可以使用二3-氯丙酸酐或者二3-氯-2-甲基丙酸酐来代替上述的酰基氯。当3-氯丙酰基氯和3-氯-2-甲基丙酰基氯二者都用于上述酰胺化过程中时,作为最终产物,获得了式(2)所示的化合物,其在同一分子内具有3-氯丙酸酰胺基团和3-氯-2-甲基丙酸酰胺基团二者。最后,可以从式(2)所示的化合物通过用碱作用于该化合物而获得(甲基)丙烯酰胺化合物1。在上面的合成方案1中,从(A)到(B)的过程优选是在0-60℃进行30分钟-8小时,从(B)到(C)的过程优选是在20-45℃进行2小时-16小时,从(C)到式(2)所示化合物的过程优选在0-30℃进行30分钟-6小时,和由式(2)所示的化合物来合成(甲基)丙烯酰胺化合物1的过程优选在0℃-60℃进行1小时-12小时。本发明的前体化合物和作为最终化合物的(甲基)丙烯酰胺化合物可以根据常规方法从反应产生的混合物中分离和收集。该化合物可以例如通过用有机溶剂萃取,使用不良溶剂结晶,使用硅胶柱色谱来进行分离。式(3)所示的化合物和衍生自该化合物的(甲基)丙烯酰胺化合物也可以通过类似于式(2)所示的化合物的情况的方式来合成,除了在上述方案1中起始材料由从甘油转化为季戊四醇。本发明的前体化合物能够容易地转化成(甲基)丙烯酰胺化合物。根据通过本发明的前体化合物作为上述合成方案所示的合成中间体的(甲基)丙烯酰胺化合物的合成反应,作为最终产物的(甲基)丙烯酰胺化合物可以以高产率获得。与通用的合成方法(其中(甲基)丙烯酰胺化合物是通过将(甲基)丙烯酰基氯或者(甲基)丙烯酸酐与胺化合物在碱性条件下反应来合成的)相比,通过本发明的前体化合物作为中间体的合成反应能够以高产率来生产(甲基)丙烯酰胺化合物。通过本发明的前体化合物所获得的(甲基)丙烯酰胺化合物用光或者热而聚合来表现出硬化性质。所以,该(甲基)丙烯酰胺化合物可以作为可自由基聚合的化合物用于各种应用中。具体地,通过本发明的式(I)所示的化合物获得的式(A)所示的化合物可以作为交联剂或者固化剂,用于光敏树脂组合物或者喷墨油墨。例如,当该化合物用于JP-A-2011-214001、JP-A-2011-248354或者JP-A-2012-32556所述的实施例时,其表现出在这些文献中所述的效果。根据本发明,可以提供用作合成(甲基)丙烯酰胺化合物的前体的化合物。本发明将基于下面的实施例来更详细地描述,但是本发明并非打算局限于此。在下面的实施例中,关于组合物的术语“份数”和“%”是质量值,除非另有所指。实施例如下来合成一种其中在式(2)中R1是H且X1是Cl(氯原子)的化合物。下文中,该化合物称作示例性化合物(1)。示例性化合物(1)示例性化合物(1)的合成在1L体积的高压釜中,放入40g的1,2,3-三(2-氰基-乙氧基)丙烷(由TokyoChemicalIndustryCo.,Ltd.制造)、100wt%的Co催化剂(RaneyCobalt2700,由W.R.Grace&Co.制造)和600mL的溶液(25%氨水:甲醇=1:1),将所形成的混合物悬浮,并且密封反应容器。将10MPa的氢引入到该反应容器中,并且在25℃的反应温度反应16小时。通过1HNMR来确认原材料的消失,该反应混合物进行Celite过滤,并且将该Celite过滤器用甲醇清洗若干次。在减压下从滤出液中蒸馏掉溶剂,因此获得中间体(C)。将所获得的中间体(C)无需进一步净化而用于接下来的反应。向装备有搅拌器的2L体积的三颈烧瓶中加入26.3gof中间体(C)、35.35g(3.5当量)的三乙胺和1L乙腈,并且在冰浴中在2小时内逐滴加入41.57g(3.3当量)的3-氯丙酰基氯,然后将所形成的混合物在室温搅拌1小时。通过1H-NMR来确认原材料的消失,在减压下从反应混合物中蒸馏掉溶剂,并且将所形成的产物进行Celite过滤,和在减压下再次蒸馏掉溶剂。最后,将所形成的产物是通过柱色谱法(乙酸乙酯:甲醇=4:1)进行净化,和因此获得在常温为白色的固体(产率:60%)。所获得的白色固体是通过1H-NMR、13C-NMR、IR和MS在下面的测量条件下鉴别的。鉴别数据表示在图1-4中。1H-NMR溶剂:氘代氯仿,内标:TMS13C-NMR溶剂:氘代氯仿,内标:TMSIR根据溴化钾(KBr)压片方法测量光谱并转化成吸光度。MS溶剂:MeOH/H2O=9/1,10mMCH3COONH4作为上述鉴别的结果,确认了所述白色固体具有示例性化合物(1)的结构。实施例2如下来合成了其中在式(3)中R1是H,和X1是Cl(氯原子)的化合物。下文中,该化合物称作示例性化合物(2)。示例性化合物(2)示例性化合物(2)的合成下面的中间体(C)是根据WO2009/154747所述的方法或者与其相当的方法来合成的。中间体(C)向装备有搅拌器的2L体积的三颈烧瓶中加入36.4g中间体(C)、48.48g(4.8当量)的三乙胺和1L的乙腈,并且在冰浴中在2小时内逐滴加入55.42g(4.4当量)的3-氯丙酰基氯,然后将所形成的混合物在室温搅拌1小时。通过1H-NMR来确认原材料的消失,在减压下从反应混合物中蒸馏掉溶剂,并且将所形成的产物进行Celite过滤,和在减压下再次蒸馏掉溶剂。最后,将所形成的产物是通过柱色谱法(乙酸乙酯:甲醇=4.5:1)进行净化,和因此获得在常温下的白色固体(产率:53%)。所获得的白色固体是通过1H-NMR、13C-NMR、IR和MS在下面的测量条件下鉴别的。鉴别数据表示在图5-8中。1H-NMR溶剂:氘代氯仿,内标:TMS13C-NMR溶剂:氘代氯仿,内标:TMSIR根据溴化钾(KBr)压片方法测量光谱并且转化成吸光度。MS溶剂:MeOH/H2O=9/1,10mMCH3COONH4作为上述鉴别的结果,确认了所述白色固体具有示例性化合物(2)的结构。参考实施例:从示例性化合物(1)或者示例性化合物(2)衍生丙烯酰胺化合物,并且评价该丙烯酰胺化合物的硬化性质。1.从示例性化合物(1)或者示例性化合物(2)合成丙烯酰胺化合物丙烯酰胺化合物(A1)和丙烯酰胺化合物(A2)是如下来获得的:根据RussianJournalofGeneralChemistry,2005,第75卷第6期,第915-922页和美国专利No.4914225所述的方法或者根据与其相当的方法,将碱作用于实施例1所获得的示例性化合物(1)上和实施例2所获得的示例性化合物(2)上。丙烯酰胺化合物(A1)丙烯酰胺化合物(A2)2.丙烯酰胺化合物(A1)和丙烯酰胺化合物(A2)的硬化性质的评价根据下面的方法评价了所获得的丙烯酰胺化合物(A1)和丙烯酰胺化合物(A2)的硬化性质(热固性)。[硬化性质的评价]制备了含有丙烯酰胺化合物(A1)或者丙烯酰胺化合物(A2)、自由基聚合引发剂和有机溶剂的样品溶液,并且将其施涂到铜板上,然后将其加热,并且进行自由基聚合,评价加热之前和之后的触感。另外,使用FT-IR(VARIAN3100FT-IR(商标名),由Varian,Inc.制造),通过由丙烯酸基团产生的在806cm-1处的峰的热降低来确认自由基聚合的进行。细节在下面给出。用于评价的样品液体1A是通过将250mg的丙烯酰胺化合物(A1)和25mg作为自由基聚合引发剂的偶氮二异丁腈(AIBN)溶解在1mL甲醇中来制备的。然后,量取10μL的这种用于评价的样品液体1A,并且将其施涂到铜板上。通过FT-IR测量其上施涂有该样品液体的铜板,并且确认了由丙烯酸基团产生的在806cm-1处的峰。然后,将该铜板在氮气氛下的100℃的烘箱中加热1小时。当加热后再次通过FT-IR来测量该铜板时,由丙烯酸基团产生的在806cm-1处的峰降低。从这个结果,证实了丙烯酰胺化合物(A1)自由基聚合的进行。此外,当评价加热之前和之后的触感时,加热后的样品板在触摸该样品板时没有感觉到发粘,和当该样品板用手指球(aballofafinger)摩擦时,与摩擦前的感觉相比,没有感觉到变化。这些结果表明了施涂到铜板上的样品液体是通过加热硬化这一事实。然后,以类似于丙烯酰胺化合物(A1)的方式评价了丙烯酰胺化合物(A2)的硬化性质。结果,丙烯酰胺化合物(A2)也被证实具有类似于丙烯酰胺化合物(A1)程度的硬化性质。已经描述了与本发明的实施方案有关的我们的发明,我们的目的是除非另有指示,否则本发明不限于说明书的任何细节,而是在附加的权利要求中阐明的它的主旨和范围内进行宽泛的解释。本申请要求2012年3月22日在日本申请的专利申请No.2012-065145的优先权,其全部在此通过引用并入。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1
- 制备N‑异丙基(甲基)丙烯酰胺的方法与制造工艺
- 一种单片氧化石墨烯/聚乙二醇单甲醚甲基丙烯酰胺水相复合材料及其制备方法与制造工艺
- 羧甲基魔芋葡甘聚糖‑纳米聚多巴胺微球的制备方法与制造工艺
- 隔震式N,N‑二甲基‑1,3‑丙二胺加氢反应釜的制造方法与工艺
- 四甲基哌啶胺的生产装置的制造方法
- 一种防滑移的核壳型三防整理剂及其制备方法
- 一种含有胺鲜酯和1?甲基环丙烯的农药组合物的制作方法
- 一种基于聚(n-异丙基丙烯酰胺)热致构象可逆转变的抗菌剂活性调用开关的制作方法
- 以羧甲基马铃薯淀粉与丙烯酰胺为原料合成吸附Fe<sup>3+</sup>树脂的方法
- 以羧甲基马铃薯淀粉与丙烯酰胺为原料合成吸附Cr<sup>3+</sup>树脂的方法
- 以羧甲基马铃薯淀粉与丙烯酰胺为原料合成吸附Pb<sup>2+</sup>树脂的方法
- 以羧甲基马铃薯淀粉与丙烯酰胺为原料合成吸附Cu<sup>2+</sup>树脂的方法
- 以羧甲基马铃薯淀粉与丙烯酰胺为原料合成吸附Ag<sup>+</sup>树脂的方法
- 丙烯酰胺系聚合物、纸力增强剂和纸的制作方法
- 使用含丙烯酰胺的过滤器分离蛋白的制作方法
- 以羧甲基马铃薯淀粉与丙烯酰胺为原料合成吸附Co<sup>2+</sup>树脂的方法
- 以羧甲基马铃薯淀粉与丙烯酰胺为原料合成吸附Ni<sup>2+</sup>树脂的方法
- 一种保水型腐植酸控释肥料及其制备方法
- 一种使富含淀粉的热加工食品增色并降低丙烯酰胺含量的方法
- 一种制备2-丙烯酰胺基-2-甲基丙磺酸用反应器的制造方法
- 以羧甲基马铃薯淀粉与丙烯酰胺为原料合成吸附Cu<sup>2+</sup>树脂的方法
- 以羧甲基马铃薯淀粉与丙烯酰胺为原料合成吸附Ag<sup>+</sup>树脂的方法
- 使用含丙烯酰胺的过滤器分离蛋白的制作方法
- 以羧甲基马铃薯淀粉与丙烯酰胺为原料合成吸附Co<sup>2+</sup>树脂的方法
- 以羧甲基马铃薯淀粉与丙烯酰胺为原料合成吸附Ni<sup>2+</sup>树脂的方法
- 丙烯酸及其酯类植绒胶及其制备方法和植绒工艺的制作方法
- 一种含羧化丙烯腈多元化物仿活性涂料印花粘合剂及其制备方法
- 一种使富含淀粉的热加工食品增色并降低丙烯酰胺含量的方法
- 一种含有1-甲基环丙烯的农药组合物的制作方法
- 一种聚醋酸乙烯胶黏剂的制作方法