含咔唑和三苯基磷/磷氧的有机电致发光器件主体材料的制作方法与工艺
本发明涉及有机电致发光技术领域,尤其涉及一类含咔唑和三苯基磷/磷氧的有机电致发光器件主体材料。
背景技术:
有机发光二极管(OLED)被认为是下一代照明和全显色技术。目前,报道的最高效率的发光器件是基于全磷光发射有机发光二极管而实现的,较高的自旋-轨道耦合效率使得磷光发射材料既可以利用单线态激子又可以利用三线态激子,从而使得内量子效率在理论上能够达到100%。但是,目前蓝光磷光发光器件来说仍然是制约有机发光二极管发展的一个瓶颈。首先,可以用于有机发光二极管的优秀蓝色磷光材料有限。目前,使用最广泛的蓝光材料为双(4,6-二氟苯基吡啶-N,C2)吡啶甲酰合铱(Ⅲ)(FIrpic,三线态能级2.62eV),但是该材料为天蓝色。其次,可作为有机发光二极管蓝色磷光的主体材料较少。蓝色磷光材料通常不是直接用于制备有机发光二极管的发射层,而是分散在主体材料中,这样才能避免三线态淬灭和浓度淬灭。这就要求主体材料的三线态能及要高于蓝光磷光材料的三线态能级才能有效的避免能量由磷光材料向主体材料回传。以目前使用最广泛的蓝光掺杂材料FIrpic来说,必须要求主体材料的三线态能级高于2.65eV才能有效的抑制能量的回传。制备有机发光二极管常用的主体材料包括小分子和高分子两类。目前,高效率有机发光二极管器件主要采用的小分子主体材料。虽然小分子有机发光二极管的效率要比高分子发光二极管的效率普遍要高,但是制备小分子有机发光二极管往往采用的是蒸镀技术,工艺复杂耗时耗能且成本较高。溶液法被认为是现阶段最适合大规模制备有机发光二极管的技术,此类技术工艺简单且成本低。但是现有的高分子主体材料以芴或咔唑作为结构单元,存在三线态能级低和载流子传输不平衡的问题。鉴于此,合成适合溶液法加工、具有高三线态能级的聚合物主体材料是解决有机发光二极管瓶颈急需解决的问题。
技术实现要素:
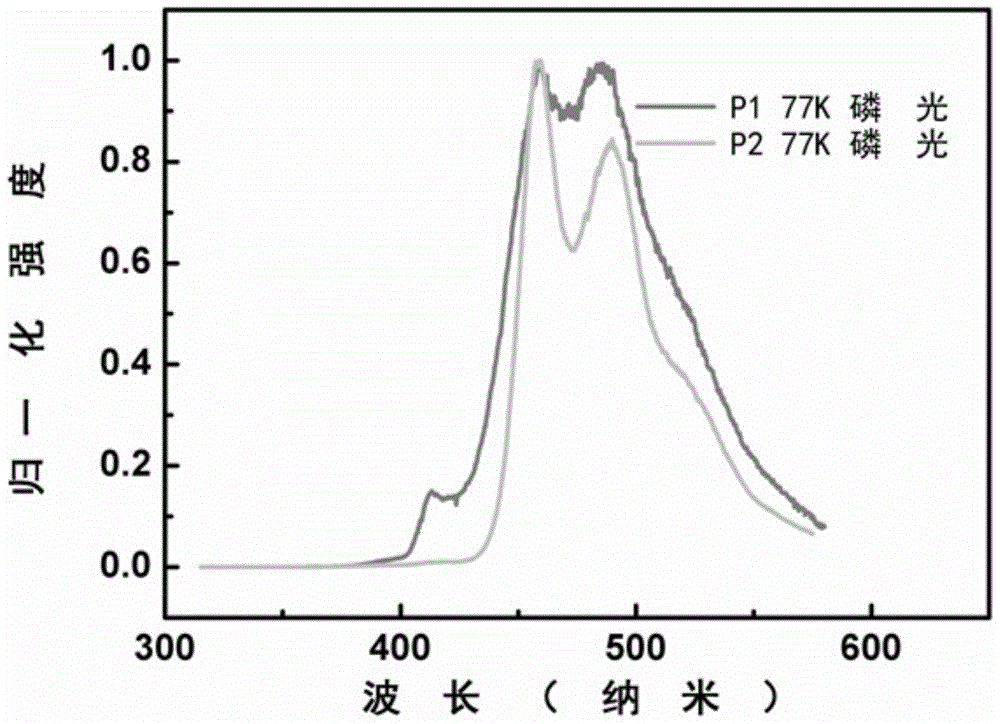
为了解决上述问题,本发明目的在于提供利于三线态能级的提高和稳定性的增加的含咔唑和三苯基磷/磷氧基团的有机电致发光器件主体材料的化合物。本发明的技术方案是:含咔唑和三苯基磷/磷氧基团的有机电致发光器件主体材料的化合物,该化合物的结构式如下:含咔唑和磷原子的有机电致发光器件主体材料的化合物,该聚合物的结构通式为:其中,式中R1为1到12个碳原子的直链或支化的烷基链,R2为苯基、噻吩基、烷基取代的苯基、噻吩基,烷基、咔唑基、烷基取代的咔唑基、芴基、烷基取代的芴基,卤原子或硼酸酯;0<x<1、0<y<1,且x+y=1;n取值为正整数。优选的,当x和y都为0.5时,所述化合物具有式P1结构:P1优选的,当x和y都为0.5时,所述化合物具有式P2结构:P2本发明的有益效果是,由于采用上述技术方案,本发明的聚合物具有以下优点:1)咔唑和三苯基磷/磷氧基团具有较高的空穴和电子传输性能,并且咔唑具有较高的三线态能级(3.01V);2)三苯基磷/磷氧基团的引入可以形成四面体结构,有效的打断高分子主链的共轭,使聚合物具有较高的三线态能级,并使高分子主链扭曲,减弱分子间π-π分子间作用,并且阻碍了分子间的堆积和结晶化,增加了分子的刚性,能有效的增加成膜后的稳定性;3)在刚性的咔唑基团9位的N上引入苯基后使得侧链与主链之间发生一定扭曲,打断侧链和主链之间的相互作用,并使得侧链和主链之间的共轭打断;4)苯基连接的烷基链可以有效的增加分子的溶解性,并且为接入其他基团提供了位点。所有这些都有利于三线态能级的提高、溶解性的改善和稳定性的增加。附图说明图1为聚合物P1和P2在二氯甲烷溶液和薄膜中的紫外吸收光谱和荧光发射光谱。图2为77K下聚合物P1和P2在2-甲基四氢呋喃中的磷光发射。图3为聚合物P1和P2的循环伏安曲线。具体实施方式下面通过具体实施例对本发明做进一步的说明,以下具体实施例为了更好的说明本发明但并不限制本发明。实施例1:M1的合成在250ml烧瓶中加入对碘苯酚(22g,100mmol)、1-溴辛烷(25.09g,130mmol)、碳酸钾(18g,130mmol)、丙酮100ml,回流下过夜反应,然后将溶剂蒸干,用二氯甲烷萃取得到的有机相用无水硫酸钠干燥,得到粘稠状液体,经过层析柱得到1-碘-4-(辛氧基)苯33g(产率99%)。1HNMR(400MHz,CDCl3)δ=7.52(d,J=8.8,2H),6.65(d,J=8.8,2H),3.88(t,J=6.5,2H),1.90–1.78(m,2H),1.41(s,2H),1.27(s,8H),0.87(t,J=6.3,3H).将3,6-二溴咔唑(10g,28mmol)、1-碘-4-(辛氧基)苯(14.4g,33mmol)、碘化亚铜(285mg,1.5mmol)磷酸三钾(6.4g,30mmol)、反式1,6-环己二胺(3mol,370ul)加入到250ml烧瓶中,加入甲苯(100mL),氩气保护下回流40小时。停止加热,冷却到室温,将溶剂蒸干,用二氯甲烷萃取,将有机相用无水硫酸钠干燥后,蒸干溶剂通过柱层析得到M112g,产率为80.7%。1HNMR(400MHz,CDCl3)δ=8.33(d,J=20.4,0.4H),8.13(d,J=18.9,1.6H),7.66(d,J=23.7,0.4H),7.50(d,J=24.2,1.6H),7.33(s,1H),7.24(s,1H),7.15(s,1H),7.07(s,1H),4.27(d,J=55.1,1H),3.96(d,J=57.7,1H),1.83(s,2H),1.49(s,1H),1.26(d,J=33.1,9H),0.87(d,J=14.8,3H)。MS(MALDI-TOF):m/zC26H27Br2NO理论值529.3;测定值529.1(M+)。反应式如下:M2的合成-78℃下,向1,4-二溴苯(14.2g,60mmol)的四氢呋喃溶液中滴加正丁基锂(1.6M的正己烷溶液)26.25mL,在氩气保护下保温1.5小时,然后滴加苯基二氯膦(5.9g,4.5mL),滴加完毕后再在-78℃下反应1.5小时,然后转移到室温下搅拌过夜。将溶剂蒸干,用二氯甲烷萃取,用无水硫酸钠干燥有机相,除去溶剂,经过层析柱得到双(4-溴苯基)(苯基)膦10g,产率79.3%。1HNMR(400MHz,CDCl3)δ=7.46(d,J=7.7,4H),7.35(d,J=4.1,3H),7.27(d,J=7.5,2H),7.13(t,J=7.6,4H).13CNMR(101MHz,CDCl3)δ=135.98,135.92,135.86,135.28,135.07,133.77,133.57,131.86,131.79,129.25,128.83,128.76,123.69。双(4-溴苯基)(苯基)膦(8g,18mmol)溶于100ml四氢呋喃中,冷却至-78,滴加正丁基锂(1.6M的正己烷溶液)20mL,在此温度下氩气保护反应1.5h,让后滴加2-异丙氧基-4,4,5,5-四甲基-1,3,2-二杂氧戊硼烷(9.8mL,45mmol),滴加完毕后,急需反应1.5h,然后生至室温反应过夜。将四氢呋喃蒸干,通过柱层析得到白色的固体M27.2g,产率77.8%。1HNMR(400MHz,CDCl3)δ=7.86(d,J=6.5,0.6H),7.73(d,J=7.6,3H),7.68–7.58(m,1H),7.55–7.47(m,0.3H),7.42(t,J=7.8,0.3H),7.31–7.25(m,7.8H),1.31(s,24H).13CNMR(101MHz,CDCl3)δ=140.65,140.53,134.64,134.58,134.04,133.85,133.01,132.82,128.87,128.56,128.45,83.81,24.81。MS(MALDI-TOF):m/zC30H37B2O4P理论值512.2;测定值513.2(M++1)。反应式如下:P1的合成将单体M1(0.50g,0.9450mmol)、单体M2(0.4860g,0.9450mmol)、碳酸钾水溶液(2M,5mL)、一滴甲基三辛基氯化铵(Aliquat336)、四三苯基膦钯(Pd(PPh3)4,21.8mg,)和20ml甲苯加入到50ml双口烧瓶中,氩气保护下,在90℃避光条件下反应48小时。然后加入苯硼酸(100mg)的脱氧甲苯溶液(5mL)和溴苯(0.5mL),分别反应12小时进行封端。冷却至室温,将混合液体导入到甲醇中,产生的沉淀经过滤后用二氯甲烷萃取,用无水硫酸钠干燥有机相,进行浓缩得到合适的体积后,用甲醇沉淀得到沉淀。沉淀收集后用在索氏提取器中分别用正己烷、甲醇抽提除去小分子和催化剂,最后用三氯甲烷抽提。得到的溶液浓缩后用三氯甲烷/甲醇沉淀,得到白色纤维状固体0.4g,产率67%。1HNMR(400MHz,CDCl3)δ=8.79–8.17(m,1H),7.67(s,13H),7.43(s,6H),7.14–6.98(m,2H),4.05(s,2H),1.83(s,2H),1.52–1.40(m,2H),1.32–1.18(m,8H),0.98(t,J=7.2,3H)。13CNMR(101MHz,CDCl3)δ=145.68,140.13,138.58,134.72,132.17,132.10,132.01,128.96,128.56,128.44,127.28,121.73,121.02,114.35,59.16,31.88,29.74,29.50,29.33,26.15,24.19,22.60,19.90,14.12,13.74.元素分析:(C44H40NOP)n理论值C83.94,H6.36,N2.22;测定值C84.35,H6.49,N1.97。反应式如下:实施例2:M1的合成在250ml烧瓶中加入对碘苯酚(22g,100mmol)、1-溴辛烷(25.09g,130mmol)、碳酸钾(18g,130mmol)、丙酮100ml,回流下过夜反应,然后将溶剂蒸干,用二氯甲烷萃取得到的有机相用无水硫酸钠干燥,得到粘稠状液体,经过层析柱得到1-碘-4-(辛氧基)苯33g(产率99%)。1HNMR(400MHz,CDCl3)δ=7.52(d,J=8.8,2H),6.65(d,J=8.8,2H),3.88(t,J=6.5,2H),1.90–1.78(m,2H),1.41(s,2H),1.27(s,8H),0.87(t,J=6.3,3H).将3,6-二溴咔唑(10g,28mmol)、1-碘-4-(辛氧基)苯(14.4g,33mmol)、碘化亚铜(285mg,1.5mmol)磷酸三钾(6.4g,30mmol)、反式1,6-环己二胺(3mol,370ul)加入到250ml烧瓶中,加入甲苯(100mL),氩气保护下回流40小时。停止加热,冷却到室温,将溶剂蒸干,用二氯甲烷萃取,将有机相用无水硫酸钠干燥后,蒸干溶剂通过柱层析得到M112g,产率为80.7%。1HNMR(400MHz,CDCl3)δ=8.33(d,J=20.4,0.4H),8.13(d,J=18.9,1.6H),7.66(d,J=23.7,0.4H),7.50(d,J=24.2,1.6H),7.33(s,1H),7.24(s,1H),7.15(s,1H),7.07(s,1H),4.27(d,J=55.1,1H),3.96(d,J=57.7,1H),1.83(s,2H),1.49(s,1H),1.26(d,J=33.1,9H),0.87(d,J=14.8,3H)。MS(MALDI-TOF):m/zC26H27Br2NO理论值529.3;测定值529.1(M+)。反应式如下:M3的合成将苯基双4-(4,4,5,5-四甲基-1,3,2-二氧硼戊环-2-基)苯基膦(M2)(5.16g,10mmol)、六水合三氯化铁(305.4mg,0.791mmol)、硫氰化铁(230.64mg,2.37mmol)、催化量的碘加入到100ml烧瓶中,加入60mL乙腈,在氧气连续鼓气下,80℃反应30分钟,完毕后冷却至室温。将溶剂蒸干,通过柱层析得到白色固体M35g,产率94%。1HNMR(400MHz,CDCl3)δ=7.85(d,J=5.7,4H),7.70–7.56(m,6H),7.53–7.47(m,1H),7.42(d,J=6.7,2H),1.32(s,24H).13CNMR(101MHz,CDCl3)δ=135.57,134.60,134.55,134.48,132.14,132.04,131.97,131.25,131.15,128.55,128.43,84.20,24.87。MS(MALDI-TOF):m/zC30H37B2O5P理论值530.2;测定值531.2(M++1)。P2的合成将单体M1(0.5293g,1.0mmol)、单体M3(0.5302g,1.0mmol)、碳酸钾水溶液(2M,5mL)、一滴甲基三辛基氯化铵(Aliquat336)、四三苯基膦钯(Pd(PPh3)4,23.1mg,)和20ml甲苯加入到50ml双口烧瓶中,氩气保护下,在90℃避光条件下反应48小时。然后加入苯硼酸(100mg)的脱氧甲苯溶液(5mL)和溴苯(0.5mL),分别反应12小时进行封端。冷却至室温,将混合液体导入到甲醇中,产生的沉淀经过滤后用二氯甲烷萃取,用无水硫酸钠干燥有机相,进行浓缩得到合适的体积后,用甲醇沉淀得到沉淀。沉淀收集后用在索氏提取器中分别用正己烷、甲醇抽提除去小分子和催化剂,最后用三氯甲烷抽提。得到的溶液浓缩后用三氯甲烷/甲醇沉淀,得到白色纤维状固体0.45g,产率69.4%。1HNMR(400MHz,CDCl3)δ=8.41(s,1H),8.28(d,J=18.7,1H),7.74(d,J=51.7,11H),7.45(dd,J=28.0,20.1,8H),7.10(s,2H),4.04(s,2H),1.83(s,2H),1.45–1.11(m,10H),0.88(s,3H)。13CNMR(101MHz,CDCl3)δ=145.60,145.35,144.08,141.51,132.91,132.71,132.13,129.04,128.67,128.50,128.29,127.36,126.98,126.00,119.11,118.15,115.81,110.31,68.29,31.88,29.38,26.15,22.68,14.25。元素分析:(C44H40NO2P)n理论值C80.12,H6.07,N2.12;测定值C,80.97;H5.86;N2.01。反应式如下:聚合物性能表征:聚合物P1和P2的光谱性质、电化学性质1)化合物P1和P2的光谱性质图1为聚合物P1和P2在二氯甲烷及薄膜中的紫外吸收和荧光发射光谱。由图1可知聚合物P1和P2的溶液中紫外吸收峰位于225nm和303nm左右,最大发射峰分别在387和393nm。薄膜中的最大吸收峰分别位于285和303nm左右,最大发射峰位于410和428nm。由P1和P2薄膜状态的吸收边缘计算得到两者带系(Eg)分别为3.18和3.26eV。图2,由77K下的磷光光谱可以得到P1和P2的三线态能级(ET)为2.72eV(三线态能级根据公式ET=1240/λ计算得到,其中λ为磷光发射光谱第一个发射峰的峰位置),聚合物P1和P2的高三线态可以满足作为有机电致发光器件主体材料的要求。2)化合物P1和P2的电化学性质图3为聚合物P1和P2的循环伏安曲线。电解池采用三电极体系,以铂碳电极为工作电极,铂丝为对电极,银/氯化银电极为参比电极,四丁基六氟磷酸铵作为支持电解质。扫描速率为100毫伏每秒。由循环伏安曲线显示聚合物P1和P2的起始氧化电位分别为1.05和1.15V,由此计算HOMO能级分别为-5.42和-5.52V(二茂铁做内标,校正电位0.43V)。依据HOMO和Eg得到LUMO分别为-2.24和-2.26eV。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1