新的重组双功能融合蛋白、其制剂和用途的制作方法
本发明涉及一种重组双功能融合蛋白、其制剂和用途,特别是在肿瘤疗法中的用途。
背景技术:
癌细胞已经发展出至少三种逃避宿主免疫监视的机制。1)通过高表达膜细胞PD-L1和PD-L2来逃避T淋巴细胞的免疫监视,其中膜细胞PD-L1和PD-L2均与T细胞表面上的PD-1结合,引起T细胞的细胞凋亡。2)逃避自然杀伤(NK)细胞的免疫监视。NK细胞表面上的NKG2D蛋白,在与癌细胞表面上的MICA/MICB蛋白结合时,可以激活NK细胞,NK细胞可以随后杀死癌细胞。然而,癌细胞已经发展出促进MICA/MICB从癌细胞上脱离的机制。脱离的MICA/MICB与NKG2D结合,阻断对NK细胞的激活。3)逃避巨噬细胞(Mφ)的免疫监视。几乎所有的癌细胞在其表面上表达高水平的CD47[1],CD47与Mφ表面上的信号调节蛋白α(SIRPα)结合,从而引发抑制性信号的生成,该抑制信性号抑制Mφ的吞噬功能[2]。有效抗癌药物的开发需要以这些机制为目标[1]。
此外,癌细胞的生长依赖于营养物的充分供给。癌细胞自身可以分泌促进血管生长的因子,例如血管表皮生长因子(VEGF)。VEGF活性的抑制将停止对肿瘤的血液供应,从而抑制癌的生长。例如,阿瓦斯汀(Avastin),FDA认证的抗体药物,起到通过抑制VEGF生物活性而治疗癌症(结肠癌、肺癌)的作用。另一蛋白药物,用于治疗结肠癌的阿柏西普(Zaltra),2012年8月批准上市,也靶向VEGF。然而,这些药物仅在某种程度上抑制癌细胞生长,而无法消除癌细胞。
信号调节蛋白是跨膜蛋白家族,具有三个成员:SIRPα(CD172a)、SIRPβ(CD172b)、SIRPγ(CD172g)。所有三个成员均包含相似的胞外区域,但是具有不同的胞内结构域。胞外结构域包含三个Ig样区域,其中第一Ig样区域为Ig-V区,而第二和第三区域为Ig-C区。
SIRPα(CD172a)的胞内结构域包含两个抑制性信号传导区域,其可以抑制信号转导以及相应的细胞功能。SIRPβ(CD172b)和SIRPγ(CD172g)的胞内结构域非常短,且不包含信号转导区域,但是SIRPβ(CD172b)可以经由衔接蛋白例如DAP12而作用于信号转导(参见图1)。SIRP主要表达在巨噬细胞(Mφ)、树突状细胞(DC)和神经元中。
CD47也是属于免疫球蛋白超家族的跨膜糖蛋白,且在包括红细胞在内的所有细胞类型的表面上表达。CD47的配体包括整联蛋白、血小板反应蛋白-1和SIRP。CD47具有很多生物功能,包括在细胞迁移、T细胞和DC激活、以及神经发育中的功能。此外,CD47通过与SIRPα相互作用,可以抑制巨噬细胞的吞噬作用。通过发出“不要攻击”信号,CD47保护正常细胞例如血细胞免受巨噬细胞的攻击。
如上所述,很多过表达CD47的肿瘤或癌细胞,通过与巨噬细胞的细胞表面上的SIRPα结合,防止巨噬细胞对癌细胞的吞噬作用。过表达CD47的癌细胞包括急性髓细胞样白血病(AML)、慢性髓细胞样白血病(CML)、急性淋巴细胞白血病(ALL)、非霍奇金淋巴瘤(NHL)、多发性骨髓瘤(MM)、膀胱癌、卵巢癌、前列腺癌、肺癌、结肠癌、乳腺癌和胰腺癌。向荷肿瘤的小鼠注射CD-47特异性抗体可以显著地抑制体内肿瘤生长[3-4]。当将同一抗体注射到携带人血癌细胞的小鼠中时,该小鼠的癌细胞完全消除[5]。
VEGF是分泌糖蛋白家族,分子量为约40kDa。该家族具有5个成员,包括VEGF-A、VEGF-B、VEGF-C、VEGF-D和PIGF。有三种VEGF受体(VEGFR),包括VEGFR1、VEGFR和VEGFR3,其各自选择性地与不同配体结合。例如,VEGFR1与VEGF-A、VEGF-B和PIGF结合,VEGFR2与VEGF-A、VEGF-C以及VEGF-D结合,而不与PIGF结合;VEGFR3仅与VEGF-C和VEGF-D结合。受体具有不同的功能。VEGFR1以非常高的亲和性与VEGF-A结合,该亲和性比VEGFR2高10倍,因而其起到负调控VEGFR2的作用。VEGFR2诱导血管表皮细胞生长,促进血管的生长。另一方面,VEGFR3与淋巴导管的发生和生长有关。在所有的VEGF及其受体中,可以说VEGF-A和VEGFR2是最重要的,因为在某些疾病状态下(例如癌症和老年性黄斑变性),VEGF-A的过度分泌,随后于与VEGFR2结合,将造成异常的血管生长,从而引起疾病发展或恶化。因而,VEGF-A和VEGFR2均为重要的药物靶点。
贝伐单抗(Bevacizumab),一种抗VEGF单克隆抗体,在2004年批准上市,并适用于转移性结肠癌、肺癌和肾癌。Zaltra,也称为VEGF-Trap,一种重组蛋白药物,也靶向VEGF,在2012年批准上市以治疗结肠癌。雷莫芦单抗(Ramucirumab),靶向VEGFR2,也正处于III期临床试验。市场上没有抗-CD47药物,所有均处于临床前阶段。
迄今没有对于任何同时靶向CD47和VEGF的单分子药物,而本发明满足该需求。
技术实现要素:
本发明公开一种重组双功能融合蛋白,其包含经由Ig的Fc片段与VEGFR的胞外结构域的Ig区连接的信号调节蛋白(SIRP)的胞外结构域的Ig区,其中,该蛋白能够同时与CD47以及VEGF结合,从而阻断CD47与巨噬细胞细胞表面上SIRP的结合以刺激巨噬细胞对肿瘤细胞的吞噬作用,并且抑制由VEGF引起的血管内皮细胞生长。本申请还提供编码该重组双功能融合蛋白的核酸分子和表达该蛋白的表达载体,用于制备该蛋白的方法,以及用于治疗过表达CD47或VEGF的疾病的方法。
在一个实施方式中,本发明的重组双功能融合蛋白包含经由接头(linker)与VEGF受体(VEGFR)的胞外结构域的Ig样区域连接的信号调节蛋白(SIRP)的胞外结构域的Ig样区域,其中,该蛋白能够与CD47以及VEGF结合,从而阻断CD47与巨噬细胞细胞表面上SIRP的结合以刺激巨噬细胞对肿瘤细胞的吞噬作用,并且抑制由VEGF引起的血管内皮细胞生长。
在一个实施方式中,在重组双功能融合蛋白中的信号调节蛋白为SIRPα,且信号调节蛋白的胞外结构域的Ig样区域为SIRPαD1。
在一个实施方式中,在重组双功能融合蛋白中的VEGFR为VEGFR1,且VEGFR1的Ig样区域为VEGFR1的胞外结构域的第二Ig区(VEGFR1D2)。
在一个实施方式中,重组双功能融合蛋白的接头是Ig的Fc片段。在一个实施方式中,Fc片段是IgG1的Fc片段。
在一个实施方式中,重组双功能融合蛋白包含经由人IgG1的Fc片段与SIRPα的胞外结构域的第一Ig区(SIRPαD1)连接的人VEGFR1的胞外结构域的第二Ig区(SIRPαD1-Fc-VEGFR1D2)。在一个特定实施方式中,本发明的重组双功能融合蛋白包含与SEQ ID NO:8为95%同一性的氨基酸序列。在一个实施方式中,本发明重组双功能融合蛋白与SEQ ID NO:8之间的氨基酸序列同一性为至少98%。在一个实施方式中,本发明重组双功能融合蛋白与SEQ ID NO:8之间的氨基酸序列同一性为至少99%。在一个实施方式中,本发明重组双功能融合蛋白包含SEQ ID NO:8的氨基酸序列。
在另一实施方式中,本发明提供编码本发明重组双功能融合蛋白的多核苷酸分子,该蛋白包含经由接头与VEGF受体(VEGFR)的胞外结构域的Ig区连接的信号调节蛋白(SIRP)的胞外结构域的Ig区,其中,该蛋白能够与CD47以及VEGF结合,从而阻断CD47与巨噬细胞细胞表面上SIRP的结合以刺激巨噬细胞对肿瘤细胞的吞噬作用,并且抑制由VEGF引起的血管内皮细胞生长。
在一个实施方式中,本发明的多核苷酸分子编码重组双功能融合蛋白,该蛋白包含SIRPα,优选为SIRPαD1。在一个实施方式中,本发明的多核苷酸分子编码重组双功能融合蛋白,该蛋白包含VEGFR1,优选为VEGFR1的胞外结构域的第二Ig区(VEGFR1D2)。
在一个实施方式中,本发明的多核苷酸分子编码重组双功能融合蛋白,该蛋白包含Ig的Fc片段作为接头,优选包含IgG1的Fc片段作为接头。
在一个实施方式中,本发明的多核苷酸分子编码重组双功能融合蛋白,该蛋白包含经由人IgG1的Fc片段与SIRPα的胞外结构域的第一Ig区(SIRPαD1)连接的人VEGFR1的胞外结构域的第二Ig区(SIRPαD1-Fc-VEGFR1D2)。在一个特定实施方式中,本发明的重组双功能融合蛋白包含与SEQ ID NO:8为95%同一性的氨基酸序列。在一个实施方式中,本发明重组双功能融合蛋白与SEQ ID NO:8之间的氨基酸序列同一性为至少98%。在一个实施方式中,本发明重组双功能融合蛋白与SEQ ID NO:8之间的氨基酸序列同一性为至少99%。在一个实施方式中,本发明重组双功能融合蛋白包含SEQ ID NO:8的氨基酸序列。
在另一实施方式中,本发明提供包含本发明多核苷酸的表达载体,该多核苷酸编码重组双功能融合蛋白。
在另一实施方式中,本发明提供包含本发明表达载体的宿主细胞。
在另一实施方式中,本发明提供药物组合物,其包含本发明的重组双功能融合蛋白以及至少一种佐剂。
在另一实施方式中,本发明提供一种用于治疗由过表达CD47或过表达VEGF、或过表达两者而造成的疾病的方法,包括对患者或受试者施用治疗有效量的本发明药物组合物。在另一实施方式中,本发明提供重组双功能融合蛋白在制备药物组合物中的用途,其中该药物组合物用于治疗由过表达CD47、或过表达VEGF、或过表达两者而造成的疾病。
在一个实施方式中,本发明的方法用于治疗选自急性髓细胞样白血病(AML)、慢性髓细胞样白血病(CML)、急性淋巴细胞白血病(ALL)、非霍奇金淋巴瘤(NHL)、多发性骨髓瘤(MM)、膀胱癌、卵巢癌、前列腺癌、肺癌、结肠癌、乳腺癌、胰腺癌和肾细胞癌中的疾病。在另一实施方式中,本发明提供用于治疗克罗恩病、过敏性哮喘和类风湿性关节炎的方法。
在一个实施方式中,根据本发明的用于治疗由增强的VEGF功能和活性造成的疾病的方法,用于治疗选自老年性黄斑病变、AMD、糖尿病视网膜病、肝纤维化和恶性血管皮内细胞瘤的疾病。
附图说明
图1为SIRP结构的示意图。
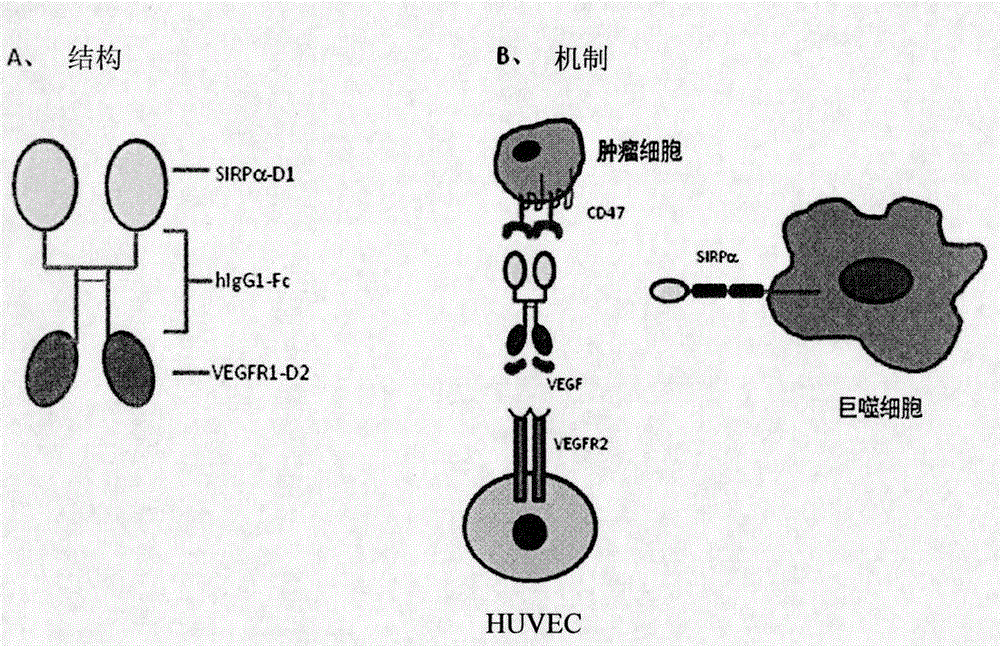
图2为SIRPαD1-Fc-VEGFR1D2的结构和作用机制的示意图。
图3示出SIRPαD1-Fc-VEGFR1D2的核酸序列(SEQ ID NO:7)和氨基酸序列(SEQ ID NO:8)。
图4示出SIRPαD1-Fc-VEGFR1D2的蛋白电泳分析。
图5示出靶标结合活性分析的结果。
图6示出靶标抑制检定的结果。
图7示出体内抗肿瘤检定的结果。
图8示出MAC-01在A549s.c.异种移植物模型中的治疗功效。分析用MAC-01处理的A组(各组n=6),并与SIRPAD1-Fc、VEGFR1D2-Fc、以及SIRPAD1-Fc加VEGFR1D2-Fc的组合的处理相比较。各分子的剂量如下:MAC-01:5mg/kg;SIRPAD1-Fc和VEGFR1D2-Fc:单用或组合时均为2.5mg/kg。当肿瘤体积达到100~200mm3时,通过腹腔注射开始处理,一周两次,持续4周。
具体实施方式
主要有三种不同的以两种或更多种肿瘤生长的药理机制为目标的途径。最常见的,可以给予患者两种或更多种不同药物的混合物。尽管该选择使得对于可能的药物组合和不同剂量有最大化的灵活度,其面临的问题为:a)由于药物负担增加以及针对各个药物的不同剂量安排,具有潜在的较差的患者依从性;b)由于药物-药物相互作用,存在可能的不相容性;以及c)药物副作用风险增加。这些问题会降低治疗的有效性并妨碍治疗目标的达成,尤其在慢性疾病例如癌症的管理中。
第二个途径依赖于对单剂型药物的固定剂量组合的使用。该途径降低药物负担,引起患者依从性的改善。固定剂量组合的不利之处主要在于,对于活性成分之间可能的剂量比的选择是有限的,这使得更加难以恰当地以最小化的不利作用将各患者的剂量调整(titrate)至最大功效。此外,组合中成分的不同药物代谢动力学特性可能在各目标患者中引起复杂的药效的时间错位,从而使总的功效折中。
第三个途径是使用在单个化合物中结合两种或更多种药理机制的多官能药物。这些多官能化合物的设计和确认较为复杂,并需要大量对于分子中靶向活性的最佳比率的研究,而联合的药物代谢动力学会在分子靶标处产生匹配的药物代谢动力学活性。多官能分子也可能面临与其他药物的固定剂量组合的检验,从而在单个药丸中组合三种或甚至四种药理机制,以产生功效的进一步增长。
经过大量实验,当前的发明者已经发明出一种新的重组双功能融合蛋白,其能够通过两个作用机制来攻击肿瘤,一个是干扰肿瘤组织的血液供给,另一个是引起巨噬细胞对肿瘤细胞的吞噬。
在一个实施方式中,本发明的重组双功能融合蛋白包含信号调节蛋白(SIRP)的胞外结构域的Ig样区域,该Ig样区域经由连接肽与VEGFR的胞外结构域的Ig样区域连接。该蛋白能够同时与CD47以及VEGF结合,从而阻断CD47与巨噬细胞细胞表面上SIRP的结合以刺激巨噬细胞对肿瘤细胞的吞噬作用,并且抑制由VEGF引起的血管内皮细胞生长。
本发明的融合蛋白包含两个靶标结合片段(例如,SIRPαD1和VEGFR1D2)以及接头(例如,Fc片段)。本领域技术人员将意识到,对于选择上述三种组分中的任意种存在很多设计选择。
接头主要作为多肽与第二异源多肽或其他类型融合多肽之间的间隔物。在一个实施方式中,接头由通过肽键连接在一起的氨基酸构成,优选为通过肽键连接的1~20个氨基酸,其中氨基酸选自20个天然存在的氨基酸。这些氨基酸中的一种或多种可以糖基化,如本领域技术人员所了解的。在一个实施方式中,1~20个氨基酸可以选自甘氨酸、丙氨酸、脯氨酸、天冬酰胺、谷氨酰胺、和赖氨酸。在一个实施方式中,接头由大部分有空键位阻的氨基酸构成,例如甘氨酸和丙氨酸。示例性的接头为多聚甘氨酸(特别是(Gly)5、(Gly)8)、多聚(Gly-Ala)、以及多聚丙氨酸。在以下实施例中示出的一个示例性合适接头为(Gly)4Ser(SEQ ID NO:X)。在另一实施方式中,本发明的双功能融合蛋白可以包含“铰链接头”,即与IgG的铰链区或部分铰链区邻接的接头序列。铰链接头序列也可以设计成改善本发明双功能融合蛋白的可制备性和稳定性。
接头也可以是非肽类接头。例如,可以使用烷基接头,例如-NH-、-(CH2)s-C(O)-,其中s=2~20。这些烷基接头还可以经任意非空间位阻基团例如低级烷基(例如C1-6)、低级酰基、卤素(例如Cl、Br)、CN、NH2、苯基等进行取代。
本发明的双功能融合蛋白也可以连接至出于赋予所需特性目的的非多肽分子,所需特性为例如减少降解和/或增加半衰期、降低毒性、降低免疫原性、和/或增加生物活性。示例性的分子包括但不限于直链多聚物例如聚乙二醇(PEG)、多聚赖氨酸、右旋糖苷;脂质;胆固醇类(例如类固醇);糖类、或寡糖分子。
例如,优选地,当对连接顺序做出设计选择时,应当将一个组分对另一组分的空间构象的影响降至最低,从而两者均保留对于它们各自靶标的高结合亲和性。连接顺序可以为SIRPαD1-Fc-VEGFR1D2、或SIRPαD1-VEGFR1D2-Fc、或VEGFR1D2-SIRPαD1-Fc。同时,普通技术人员将意识到,重组双功能融合蛋白的分子量应当最小化,以促进融合蛋白的产生,只要不损害靶标结合亲和性。
例如,接头蛋白片段可以是Fc片段、或其他合适的接头蛋白。Fc片段的大小可以为232个氨基酸,包含在铰链区的半胱氨酸、在CH2区的两个半胱氨酸、和在CH3区的两个半胱氨酸。在铰链区的半胱氨酸起到在两个单体之间形成二硫键的作用,从而生成同质二聚体,而在CH2和CH3区的半胱氨酸可以形成链内二硫键以稳定蛋白。
免疫球蛋白包括IgG、IgA、IgM、IgD和IgA,在这之中,IgG量最多且相对稳定。在本发明中优选IgG,因为其Fc片段对葡萄球菌A蛋白表现出最高的结合活性,因而可以容易地进行纯化。
例如,SIPR胞外区域的能够结合CD47的所有Ig区(SIPRα、SIPRγ)均可以用在融合蛋白中。对于VEGFR胞外区域的Ig样区域也是如此。已知VEGFR1的D2以最高活性与VEGF结合,因而在本发明中优选。
优选地,人来源的序列用在人癌治疗中,因为来自非人动物的蛋白或肽的强免疫原性可能引起过敏和其他不利作用。然而,基于不同的应用目的,其他动物蛋白或肽也可以用在本发明中。
在一个实施方式中,在本发明重组双功能融合蛋白中的信号调节蛋白为SIRPα。该信号调节蛋白的胞外结构域的Ig样区域为SIRPαD1。
在一个实施方式中,在本发明重组双功能融合蛋白中的VEGFR为VEGFR1,且VEGFR1的Ig样区域为VEGFR1胞外结构域的第二Ig样区域(VEGFR1D2)。
在一个实施方式中,在本发明重组双功能融合蛋白中的Fc片段为IgG1的Fc片段。
在一个实施方式中,本发明的重组双功能融合蛋白包含人SIRPα胞外结构域的第一Ig样区域,其经由人IgG1的Fc片段与VEGFR1胞外结构域的第二Ig样区域(VEGFR1D2)连接,从而生成SIRPαD1-Fc-VEGFR1D2。
在一个实施方式中,本发明重组双功能融合蛋白的氨基酸序列在图3B(SEQ ID NO:8)中示出。在一个实施方式中,双功能重组融合蛋白包含具有SEQ ID NO:8所列序列的多肽。在另一实施方式中,多肽的氨基酸序列与上述多肽具有至少80%、85%、90%、95%、98%或99%同一性,其中该多肽能够与CD47和VEGF两者结合且能够抑制肿瘤细胞生长。
在另一方面,本发明提供分离的核酸分子,该核酸分子包含编码双功能重组融合蛋白的多核苷酸,该双功能重组融合蛋白包含由SEQ ID NO:8所列序列的多肽。在另一实施方式中,多肽的氨基酸序列与上述多肽具有至少80%、85%、90%、95%、98%或99%同一性,其中该多肽能够与CD47和VEGF两者结合,并且能够抑制肿瘤细胞生长。
本发明还公开一种药物组合物,其包含上述的重组双功能融合蛋白、以及至少一种药学可接受的赋形剂。如果需要的话,一种或多种药学可接受的载体或赋形剂也可以包含在药物组合物中。载体包括药学中常见的稀释剂、媒介(vehicle)、填充剂、粘结剂、润湿剂、崩解剂、吸收促进剂、表面活性剂、吸附载体、润滑剂等。
这样的组合物包含治疗上或预防上有效量的多肽或蛋白,其与药学上可接受的材料和生理上可接受的制剂材料掺和在一起。药物组合物可以包含用于改变、保持或保存例如组合物的pH、渗透压、粘性、透明度、颜色、等渗性、气味、无菌性、稳定性、溶解或释放速率、吸附性或渗透性的制剂材料。合适的制剂材料包括但不限于氨基酸(例如甘氨酸、谷氨酰胺、天冬酰胺、精氨酸、或赖氨酸);抗微生物剂;抗氧化剂(例如抗坏血酸、亚硫酸钠或亚硫酸氢钠);缓冲剂(例如硼酸盐、碳酸氢盐、Tris-HCl、柠檬酸盐、磷酸盐、其他有机酸);填充剂(例如甘露醇或甘氨酸);螯合剂(例如乙二胺四乙酸(EDTA));络合剂(例如咖啡因、聚乙烯吡咯烷酮、β-环糊精或羟丙基-β-环糊精);填料;单糖;二糖和其他糖类(例如葡萄糖、甘露糖或右旋糖);蛋白(例如血清白蛋白、白明胶或免疫球蛋白);着色剂;矫味剂和稀释剂;乳化剂;亲水聚合物(例如聚乙烯吡咯烷酮);低分子量多肽;成盐平衡离子(例如钠);防腐剂(例如苯扎氯铵、苯甲酸、水杨酸、硫汞撒、苯乙醇、对羟基苯甲酸甲酯、对羟基苯甲酸丙酯、氯己定、山梨酸或过氧化氢);溶剂(例如丙三醇、丙二醇或聚乙二醇);糖醇(例如甘露醇或山梨醇);混悬剂;表面活性剂或润湿剂(例如普兰尼克、PEG、山梨醇酯、聚山梨醇酯例如聚山梨醇酯20、聚山梨醇酯80、triton、氨基丁三醇、卵磷脂、胆固醇、tyloxapal);稳定增强剂(蔗糖或山梨醇);紧张度增强剂(例如碱金属卤化物(优选为氯化钠或氯化钾)、甘露醇、山梨糖醇);递送载体;稀释剂;赋形剂和/或药物佐剂。(Remington′s Pharmaceutical Sciences,18th Edition,A.R.Gennaro,ed.,Mack Publishing Company,1990)
最佳的药物组合物将由本领域技术人员基于例如意向的给药途径、递送形式和所期望的剂量而确定。参见,例如,Remington′s Pharmaceutical Sciences,同上。这样的组合物可能会影响多肽的物理状态、稳定性、体内释放速率、以及体内清除速率。例如,合适的组合物可以是注射用水、非肠道给药用的生理盐水溶液。
在药物组合物中的主要媒介或载体的性质可以是水性或非水性的。例如,合适的媒介或载体可以是注射用水、生理盐水溶液或人工脑脊液,可能补充有常用在肠道外给药组合物中的其他材料。中性缓冲盐水或与血清白蛋白混合的盐水是另外的示例性媒介。其他示例性的药物组合物包含Tris缓冲液、或乙酸盐缓冲液,其还可以包含山梨糖醇或其合适的替代物。在本发明的一个实施方式中,组合物可以通过将具有所需纯度的所选组合物与任选的配制剂(Remington′s Pharmaceutical Sciences,同上)以冻干的块状物或水溶液的形式进行混合而制备,并存储。此外,治疗组合物可以使用合适的赋形剂例如蔗糖配制为冻干物。
制剂可以以多种方法进行递送,例如,通过吸入疗法、经口、或通过注射。当涉及肠道外给药时,用在本发明中的治疗组合物可以处于无热源、肠道外可接受的水溶液的形式,其包含在药学可接受载体中的所需多肽。用于肠道外注射的特别合适的载体为合适保存的灭菌蒸馏水,在其中,多肽配制为无菌等渗溶液。另一制剂可以涉及所需分子与作用剂的制剂,作用剂为例如可注射微球、可生物蚀解的颗粒、高分子化合物(聚乳酸、聚乙醇酸)、珠子或脂质体,这些作用剂提供用于产品的受控的或持续的释放,产品可以在之后通过积存注射而递送。也可以使用透明质酸,且其可以具有提升在循环中的持续期的效果。用于导入所需分子的其他合适手段包括可植入药物递送装置。
在另一方面,适合于注射给药的药物制剂可以配制在水溶液中,优选配制在生理上相容的缓冲液例如汉克斯液、林格氏液,或生理缓冲盐水中。水性注射混悬液可以包含提高混悬液粘度的物质,例如羧甲基纤维素钠、山梨糖醇或右旋糖。此外,活性化合物的混悬液可以制备为合适的油性注射混悬液。合适的亲脂性溶剂或载体包括脂肪油,例如脂肪油,或合成的脂肪酸酯,例如油酸乙酯、甘油三酯或脂质体。非脂质多聚阳离子型氨聚合物也可以用于递送。任选地,混悬液也可以包含合适的稳定剂或增加化合物溶解度并使得可以制备高度浓缩溶液的作用剂。在另一个实施方式中,药物组合物可以配制用于吸入。吸入溶液也可以与用于喷雾递送的推进物一起配制。在另一实施方式中,溶液可以成为喷雾状。肺给药还在PCT申请PCT/US94/001875中有过描述,该申请描述化学修饰蛋白的肺递送。
另外还关注的是,某些制剂可以经口给药。在本发明的一个实施方式中,以该方式给药的分子可以与常用在固体剂型例如片剂和胶囊剂的调和中的那些载体一起配制,或者不与载体一起配制。例如,胶囊可以设计成,在胃肠道中在生物可得性最大化且全身前降解最小化时的时点释放制剂的活性部分。可以包括其他的作用剂来促进治疗分子的吸收。也可以采用稀释剂、矫味剂、低熔点蜡、植物油、润滑剂、混悬剂、片剂崩解剂和粘结剂。用于经口给药的药物组合物也可以使用本领域熟知的药学上可接受载体以适于经口给药的剂型进行配制。这样的载体使得药物组合物可以配制为片剂、药丸、糖衣丸、胶囊剂、液体剂、凝胶剂、糖浆、浆料、混悬剂等,用于患者摄取。
经口使用的药物制剂的获得可以通过将活性化合物与固体赋形剂结合并加工所得的颗粒混合物(任选地,在磨碎后)以获得片剂或糖衣丸核部。可以添加合适的助剂,如果如所期望的。合适的赋形剂包括糖类或蛋白填料,例如糖,包括乳糖、蔗糖、甘露醇和山梨糖醇;来自玉米、小麦、水稻、土豆或其他植物的淀粉;纤维素,例如甲基纤维素、羟丙基甲基纤维素、或羧甲基纤维素钠;黏胶,包括阿拉伯树胶和黄芪胶;和蛋白,例如白明胶和胶原蛋白。如果需要的话,可以加入崩解剂或助溶剂,例如交联的聚乙烯吡咯烷酮、琼脂,以及海藻酸或其盐例如海藻酸钠。
糖衣丸的核部可以与合适的包衣联合使用,例如浓缩的糖溶液,其也可以包含阿拉伯树胶、滑石、聚乙烯吡咯烷、卡伯姆胶、聚乙二醇、和/或二氧化钛、漆溶液、以及合适的有机溶剂或溶剂混合物。染料或色素可以加至片剂或糖衣丸包衣,以用于产品识别或表征活性化合物的量(即,剂型)。
可以经口使用的药物制剂还包括由白明胶制成的硬胶囊,以及由白明胶和包衣例如丙三醇或山梨糖醇制成的密封软胶囊。硬胶囊可以包含与填料或粘结剂例如乳糖或淀粉,润滑剂例如滑石或硬脂酸镁,以及任选的稳定剂相混合的活性成分。在软胶囊中,活性化合物可以溶解或混悬在合适的液体中,例如脂肪油、液体或液体聚乙二醇,具有或不具有稳定剂。
其他药物组合物对于本领域技术人员是显而易见的,包括涉及多肽在持续递送或受控递送制剂中的制剂。用于配制多种其他持续递送或受控递送手段例如脂质体载体、生物蚀解微粒或多孔珠子和积存注射的技术也为本领域技术人员所知。参见,例如,PCT/US93/00829,其描述了多孔聚合微粒的受控释放,以递送药物组合物。持续释放制剂的其他例子包括处于成形物品形式的半透式聚合物基质,例如膜或微囊。持续释放基质可以包括聚酯、水凝胶、聚交酯(美国专利3773919,EP58481)、L-谷氨酸和γ-乙基-L-谷氨酸酯的共聚物(Sidman等人,Biopolymers,22:547-556(1983))、聚(2-羟乙基-甲基丙烯酸酯)(Langer等人,J.Biomed.Mater.Res.,15:167-277,(1981);Langer等人,Chem.Tech.,12:98-105(1982))、乙烯-醋酸乙烯(Langer等人,同上)或聚-D(-)-3-羟基丁酸(EP133988)。持续释放组合物也包含脂质体,其可以通过多种本领域已知方法中的任意种进行制备。参见,例如,Eppstein等人,PNAS(USA),82:3688(1985);EP 36676;EP 88046;EP 143949。
用于体内给药的药物组合物通常必须是无菌的。这可以通过经由无菌过滤膜的过滤而完成。在组合物为冻干的情况下,使用该方法的灭菌可以在冻干和重构前或之后进行。用于肠道外给药的组合物可以以冻干形式存储,或存储在溶液中。此外,肠道外组合物通常放置到具有无菌接达口的容器中,例如具有通过皮下注射针刺穿的塞子的静脉溶液袋或药瓶。
药物组合物一旦配制好,则可以在无菌药瓶中存储为溶液、混悬液、凝胶、乳化液、固体、或脱水或冻干的粉剂。这样的制剂可以以即用型或需要在给药前重构的形式(例如,冻干的)存储。
在特定实施方式中,本发明涉及用于制备单剂型给药单元的试剂盒。该试剂盒可以各自包含具有干燥蛋白的第一容器和具有水性配方的第二容器。还包括在本发明范围内的是一种试剂盒,其含有预先填充的单隔室或多隔室的注射器(例如,液体注射器和冻干注射器)。
治疗中采用的药物组合物的有效量将取决于例如治疗背景和目标。本领域技术人员将意识到,用于治疗的合适剂量水平将因而部分地取决于递送的分子、正在使用多肽的指征、给药途径、以及患者的身材大小(体重、身体表面或器官大小)和状况(年龄和基本健康状况)而变化。因而,临床医生可以调整剂量并修改给药途径来获得最佳的疗效。取决于上述因素,典型的剂量可以为约0.1mg/kg至最高约100mg/kg或更高。多肽组合物可以优选地静脉注射或施用。长期作用的药物组合物可以每三到四天、每周或每两周进行施用,取决于特定制剂的半衰期和清除速率。给药的频率将取决于所使用制剂中多肽的药物代谢动力学参数。典型地,施用组合物,直至剂量达到所期望的效果。因而,组合物可以以单次给药或随时间推移的多次给药(以相同或不同浓度/剂量)、或以连续输注进行给药。合适剂量的进一步改良可常规做出。合适的剂量可以通过使用合适的剂量-反应数据而确定。
药物组合物的给药途径为根据已知的方法,例如,经口地、通过静脉注射、腹膜内、脑内(脑实质内)、脑室内、肌内、眼内、动脉内、门静脉(intraportal)、病灶内途径、髓内、鞘内、心室内、经皮、皮下或腹膜内;以及鼻内、肠内、局部、舌下、尿道、阴道或直肠手段,通过持续释放系统或通过植入装置。在需要的情况下,组合物可以通过快速浓注或持续地通过输注、或通过植入装置而给药。或者,或另外地,组合物可以在局部经由植入其上吸附或包覆有所需分子的膜、海绵状物或另一合适材料而给药。在使用植入装置的情况下,装置可以植入到任何适当的组织或器官中,且所需分子的递送可以经由扩散、定时释放的大丸药、或持续的给药。
在一些情况下,本发明的双功能融合蛋白可以通过植入经使用例如本文所述的方法而遗传改造成表达并分泌多肽的某些细胞而进行递送。这样的细胞可以是动物或人细胞,且可以是自体、异源或异种的。任选地,细胞可以是无限增殖的。为降低免疫应答的几率,细胞可以进行包囊,以避免周边组织的渗透。包囊的材料通常为生物相容的、半渗聚合围绕物或膜,使得多肽产物可以释放但是防止细胞被患者免疫系统或其他来自周边组织的不利因素而破坏。
还展望体内基因疗法,其中将编码本发明双功能融合蛋白的核酸分子或其衍生物直接引入受试者中。例如,将编码本发明双功能融合蛋白的核酸序列经由带有或不带有合适递送载体例如腺相关病毒载体的核酸构建体经局部注射而引入目标细胞。可供选择的病毒载体包括但不限于逆转录病毒、腺病毒、单纯疱疹病毒、和乳头状瘤病毒载体。病毒载体的物理转移可以通过所需核酸构建体的局部注射、或包含所需核酸序列的其他合适递送载体、脂质体介导的转移、直接注射(裸露的DNA)、或微粒轰击(基因枪)而在体内实现。
本公开的组合物可以单独使用,或者与其他用于增强其疗效或降低潜在副作用的治疗剂联合使用。
本发明的另一目的是提供制备上述重组双功能融合蛋白以及包含该蛋白的药物组合物的方法。在一个实施方式中,制备方法包括以下步骤:(1)提供编码多核苷酸分子;(2)构建包含(1)的多核苷酸分子的表达载体;(3)用(2)中的表达载体转染或转化合适的宿主细胞并进行培养以在宿主细胞中表达蛋白;以及(4)纯化蛋白。该制备可以由普通技术人员用经过批准并熟知的技术进行执行。
本发明的另一目标是提供使用本发明药物组合物来治疗癌症的方法,包括向有该需求的患者或受试者施用有效量的上述药物组合物。在一个实施方式中,药物组合物用于治疗过表达CD47的肿瘤或癌症,包括但不限于急性髓细胞样白血病(AML)、慢性髓细胞样白血病(CML)、急性淋巴细胞白血病(ALL)、非霍奇金淋巴瘤(NHL)、多发性骨髓瘤(MM)、膀胱癌、卵巢癌、前列腺癌、肺癌、结肠癌、乳腺癌、胰腺癌和肾癌。
在一个实施方式中,药物组合物可以用于治疗其他相关的过表达CD47的症状,包括但不限于克罗恩病、过敏性哮喘和类风湿性关节炎。
在一个实施方式中,药物组合物可以用在需要抑制VEGF的功能或活性的疾病中,包括但不限于老年性黄斑病变(AMD)、糖尿病视网膜病(DR)、肝纤维化、恶性血管皮内细胞瘤等。
同时,本发明提供编码重组双功能融合蛋白的多核苷酸分子和表达重组双功能融合蛋白的表达载体。载体的例子包括但不限于质粒、病毒载体、酵母人工染色体(YAC)、细菌人工染色体(BAC)、可转化人工染色体(TAC)、哺乳动物人工染色体(MAC)和人工附加染色体(HAEC)。
本发明提供包含上述表达载体的宿主细胞。宿主细胞可以用表达载体进行转化或转染。合适的宿主细胞包括原核细胞、酵母和其他真核细胞。优选地,使用大肠杆菌(Escherichia coli)、酵母或哺乳动物细胞系(例如COS或CHO)。
本发明的重组双功能融合蛋白(SIRPαD1-Fc-VEGFR1D2)包含人信号调节蛋白胞外结构域的第一Ig样区域(SIRPαD1)、人VEGFR1胞外结构域的第二Ig样区域(VEGFR1D2)、以及人IgG1的Fc片段。蛋白为同质二聚体,分子量为100kDa。中国仓鼠卵巢(CHO)细胞的稳定表达细胞系通过筛选而获得,并且获得100毫克的融合蛋白。在体内证实,蛋白能够同时与CD47和VEGF结合,阻断CD47与巨噬细胞细胞表面上SIRP的结合,促进巨噬细胞对肿瘤细胞的吞噬作用,并且抑制由VEGF引起的血管内皮细胞生长。SIRPαD1-Fc-VEGFR1D2在体内测试中表现出对CD47阳性肿瘤细胞HL60的强烈功效,并且可以完全消除肿瘤。这通过抑制肿瘤组织中的血管再生并刺激巨噬细胞的吞噬作用而完成。
本发明涉及两个关键的癌靶标,SIRPα和VEGF。尽管阻断SIRPα可以促进巨噬细胞(Mφ)对肿瘤细胞的吞噬,但是大的肿瘤由于充分的血液供给而不能被有效地吞噬。单独阻断VEGF也无法消除肿瘤。当SIRPα和VEGF同时被阻断时,肿瘤在生长受阻的情况下对巨噬细胞(Mφ)的吞噬作用变得更加易感,且可以完全消除。
本发明将参照以下非限制性实施例进行进一步说明。
实施例
实施例1:SIRPαD1-Fc-VEGFR1D2与CD47以及VEGF结合,并抑制HL60肿瘤生长
1.SIRPαD1-Fc-VEGFR1D2表达载体的构建
采用Plg-Tail(R&D Systems)。在克隆前,对Plg-Tail进行遗传改造,通过用引物1和2扩增Fc编码序列、使在Fc羧基端的终止密码子缺失、并将PCR产物克隆到Plg-Tail的EcoR1/XhoI位点。用于SIRPαD1的编码序列用引物3和4从THP-1(TIB-202TM)细胞中扩增,且用于VEGFR1D2的编码序列用引物5和6从HUVEC(PCS-100-010TM)细胞中扩增。将两个PCR产物分别克隆到遗传改造的plg-Tail载体的HindIII/EcoR1和XhoI/XbaI位点,从而生成载体pSIRPαD1-Fc-VEGFR1D2。
表1PCR引物
备注:基因特异性序列以粗体示出,且限制性内切酶识别位点以下划线标记。
2.蛋白表达和纯化
将包含CHO细胞的完全细胞培养基DMEM(10%FBS)加到24孔板中,每孔0.5ml,并将板在孵育器中保持24小时。对于转染,将0.5μg质粒DNA和2μl lipofectamine 2000(Cat#11668-027,Invitrogen)分别溶解在50μl无血清培养基中,并在室温下混合20分钟,慢慢地加入孔中,并放回孵育器24小时。第二天,取100μl血清用于蛋白表达测试,通过酶联免疫吸附测定(ELISA)。
3.蛋白表达测试
通过ELISA,用以下步骤进行蛋白表达测试:将山羊抗人IgG抗体F(ab’)2片段(Biosource International Inc)溶解在PBS磷酸缓冲液中,之后加到96孔ELISA板中,每孔20ng。将ELISA板放置在冰箱中,4℃过夜。对于测试,将板用封闭液(PBS,0.05%吐温20,3%脱脂牛奶)封闭1小时,之后加入稀释血清,并在室温下孵育1小时。在用洗涤液(PBS,0.05%吐温20)洗涤5次后,加入马辣根过氧化酶(HRP)标记的兔抗人IgG抗体(Jackson ImmunoResearch Lab),并将板在室温下孵育1小时。在洗涤5次后,加入HRP的底物,并在2分钟后用终止液(1N H2SO4)终止发色反应。在450nm测量光密度。
4.筛选稳定表达的细胞系
使转染的细胞进行浓度递增的抗生素加压筛选(Geneticin,Cat#10131035,Invitrogen)。逐渐杀灭不稳定的细胞,将存活的细胞在稀释后加入到五个96孔板中,每孔0.5~1个细胞。将板放置在孵育器中10~15天。通过ELISA来测试含有单个克隆的孔,之后增殖阳性细胞,并用无血清Ex-CELL CD CHO培养基(Cat#14361C-1000ML,SIGMA)进行惯常培养。在进一步筛选后,选择呈现出高表达水平的细胞,并冷冻以备用。
5.蛋白生成和纯化
将稳定表达的细胞系(3×105/ml)接种到包含300ml无血清培养基的2L摇瓶中,并将摇瓶放置在摇床上进行培养。在细胞密度达到5×106/ml时,获得上清液。将上清液用Protein A柱纯化。将纯化的蛋白替换到透析的PBS(pH 7.0)中。采用蛋白电泳分析来确保至少98%的纯度。
6.靶标结合亲和度
SIRPαD1-Fc-VEGFR1D2对靶标CD47以及VEGF的结合亲和度通过使用流式细胞术和ELISA法进行测定。
SEM-K2细胞和HL60细胞用在结合亲和度测试中(前者为急性淋巴细胞白血病细胞,且后者为早幼粒细胞白血病细胞)。在用PBS洗涤后,将细胞以1×106/ml的浓度悬浮在PBS中。将hlgG(1μg/ml)加入到细胞悬浮液中,并将悬浮液在冰箱中4℃孵育1小时。将细胞转移到用PBS洗涤(100μl每孔)后的96孔U形细胞板中(Cat#163320,NuncTM)。之后,加入不同浓度的SIRPαD1-Fc-VEGFR1D2,并将细胞在冰箱中4℃孵育1小时。在用PBS洗涤后,使细胞悬浮,并与FITC标记的人IgG-Fc抗体(Cat#F9512,sigma)一起孵育。1小时后,通过流式细胞仪(Guava easyCyte 6HT-2L,Millipore)测试细胞。
以下列步骤,通过ELISA测试对VEGF-A的结合亲和性:
用包被缓冲液CBS(Sigma-Aldrich Co.,产品编号:1001329288C3041-100CAP)将VEGF-165稀释到1000ng/ml;向ELISA板(Cat#442404,NuncTM)加入100μl溶液(每孔100μg);将包被的板放置在冰箱中4℃过夜;在测试中,用0.05%的PBS-T洗涤该包被板一次,并然后用3%的脱脂奶对该包被板进行封闭1小时;向包被板加入稀释的SIRPαD1-Fc-VEGFR1D2(20、10、5......0.01nM)(每孔100μl);在室温下孵育1小时后,弃样品,并用0.05%的PBS-T溶液洗涤5次;添加100μl稀释的HRP-兔抗人IgG Fc(1∶20000)(Cat#:309-036-008,Jackson ImmunoResearch Lab);在室温下将溶液孵育1小时;用洗涤液将板洗涤5次;添加HRP底物;避光进行发色反应10~20分钟并通过使用1N H2SO4来终止反应;在微板酶标仪上获得OD450值。
7.靶标活性阻断检定
为测试SIRPαD1-Fc-VEGFR1D2是否能够阻断由CD47-SIRPα结合而引起的吞噬抑制性活性,将FITCCFSE标记的SEM-K2细胞与RAW264.7细胞以2∶1的RAW264.7:SEM-K2比率混合;之后在6个新的细胞培养板上将该混合物与不同浓度的SIRPαD1-Fc-VEGFR1D2、SIRPαD1-Fc以及人IgG-Fc一起培养;4小时后,小心地摇晃板,并吸取悬浮的SEM-K2细胞,用PBS将板洗涤两次来除去所有悬浮的细胞;用胰蛋白酶-EDTA来消化板壁上的RAW264.7细胞,并用PBS将板洗涤两次;经由流式细胞术来定量地分析RAW264.7细胞对SEM-K2细胞的吞噬比例。
8.抗肿瘤检定
在HL-60皮下肿瘤模型中研究SIRPαD1-Fc-VEGFR1D2的体内抗肿瘤活性。HL-60细胞系在体外人白血病模型中有较好的表征,得自于患有急性早幼粒细胞白血病的病人[6],且已经广泛地用在用于药物治疗效用的异种移植小鼠模型中[7-9]。二十(20)个Balb/c裸鼠皮下注射白血病(HL60)细胞(每只小鼠4×106个细胞)。当肿瘤生长至100~150mm3时,将小鼠分成4组:第一组为腹膜内注射PBS;第二组腹膜注射VEGFR1D2-Fc;第三组腹膜内注射SIRPαD1-Fc;第四组腹膜内注射SIRPαD1-Fc-VEGFR1D2。各个组的剂量为3mg/kg,且一周给药两次,连续6次。肿瘤的体积和重量一周测量两次。
实验结果
1.SIRPαD1-Fc-VEGFR1D2表达载体的构建
SIRPαD1-Fc-VEGFR1D2是重组Fc融合蛋白,由SIRPα的第一胞外结构域(D1)和经由人IgG1的Fc片段连接的VEGFR1的第二胞外结构域(D2)构成,称为MAC-01。其具有如图2A所示的结构。SIRPαD1和VEGFR1D2通过细胞克隆分别与N端和C端结合。SIRPαD1-Fc-VEGFR1D2的序列包含1503个核苷酸(图3A),其中信号肽编码序列具有63个核苷酸(红色),SIRPαD1具有426个核苷酸(蓝色),IgG1-Fc具有696个核苷酸(黑色),VEGFR1D2具有306个核苷酸(绿色),EcoRI序列具有6个核苷酸,且XhoI序列具有6个核苷酸。图3B示出相应的氨基酸序列。
2.蛋白表达分析
通过使用蛋白电泳(SDS-PAGE),发现蛋白分子重量在非还原条件下大于170kDa(图4),与理论值(100kDa)较为不同。这可能是因为蛋白糖基化。
3.SIRPαD1-Fc-VEGFR1D2的靶标结合亲和性
通过使用流式细胞术和ELISA,分析SIRPαD1-Fc-VEGFR1D2对VEGF-A的结合亲和性。结果显示,SIRPαD1-Fc-VEGFR1D2取向CD47(图5A)和VEGF-A(图5B)两者,EC50分别为0.1~1.0nM(CD47)和0.08~0.15nM(VEGF-A)。
4.SIRPαD1-Fc-VEGFR1D2促进巨噬细胞对肿瘤细胞的吞噬
通过使用标记的急性淋巴细胞白血病细胞SEM-K2和小鼠巨噬细胞(RAW264.7),分析SIRPαD1-Fc-VEGFR1D2对吞噬肿瘤细胞的作用。结果示出(图6),SIRPαD1-Fc-VEGFR1D2显著增强巨噬细胞对肿瘤细胞的吞噬。效果为剂量依赖型的。
5.SIRPαD1-Fc-VEGFR1D2蛋白的抗肿瘤活性
通过使用HL60皮下肿瘤模型,研究SIRPαD1-Fc-VEGFR1D2蛋白的体内抗肿瘤活性。如图7所示,VEGFR1D2-Fc处理组显示出对于肿瘤生长的非常有限的抑制作用,而SIRPαD1-Fc处理组显示出对肿瘤生长的明显的抑制作用而不足以消除肿瘤。相反,当用SIRPαD1-Fc-VEGFR1D2处理之后,小鼠显示出,肿瘤大小由处理之初的100mm3减少,且肿瘤在治疗结束时几乎消失。在阴性对照组,小鼠的肿瘤体积随时间逐渐生长,并在最后达到1000mm3。实验结果显示,仅仅抑制VEGF的活性对HL60肿瘤没有显著的治疗效果,表明HL60肿瘤的生长不在很大程度上依赖于VEGF。阻断由肿瘤引起的对巨噬细胞的吞噬作用的抑制,加强巨噬细胞对肿瘤细胞的吞噬,从而显著地抑制肿瘤细胞的生长。然而,如果VEGF和CD47-SIRPα的功能均被抑制,肿瘤将被完全消除。
上述数据表明,重组双功能融合蛋白SIRPαD1-Fc-VEGFR1D2是具有良好的双重靶标结合亲和性的新的重组蛋白。体内实验示出,SIRPαD1-Fc-VEGFR1D2蛋白具有显著的抗肿瘤活性。在HL60模型中,肿瘤可以完全消除。蛋白主要具有两种消除肿瘤的机制:抑制肿瘤中的血管生长,从而停止肿瘤生长所需的营养供给;肿瘤细胞表面的CD47与巨噬细胞表面SIRPα结合,从而破坏由肿瘤引起的自保护机制,以引发巨噬细胞对肿瘤细胞的强烈吞噬作用。这两个机制是协同的,即,肿瘤体积由于缺乏营养而减少,且因而肿瘤更加容易被巨噬细胞吞噬,引起肿瘤的消除。
实施例2MAC-01抑制肺癌(A549)生长
上述重组Fc融合蛋白SIRPαD1-Fc-VEGFR1D2(MAC-01)在A549肺癌异种移植模型上进行测试。A549细胞系最先在1972年由D.J.Gard等人通过移除一名58岁高加索男性的移植肿瘤并培养肿瘤中的癌性肺组织而开发出[10][11]。A549异种移植小鼠模型已经大量地用于很多种候选药物的效能评估[12-15]。实验数据表明,MAC-01可以同时与VEGF和CD47结合,因而能够阻断各自的分子介导的信号。当在体内肺癌异种移植模型中研究中,MAC-01表现出明显优于单个分子组合的较强抗肿瘤活性。
Balb/c裸小鼠皮下注射在200μl无血清培养基/基质胶(50∶50v/v)中的5×106个A549细胞。当平均肿瘤体积达到100~200mm3时,将30只荷有合适大小的肿瘤的小鼠根据肿瘤体积随机分成5组(每组6只小鼠)。小鼠用5mg/kg的MAC-01、或2.5mg/kg的SIRPAD1-Fc、或2.5mg/kg的VEGFR1D2-Fc、或2.5mg/kg的SIRPAD1-Fc与2.5mg/kg的VEGFR1D2-Fc的组合经i.p.处理,一周两次,持续4周。PBS处理用作阴性对照。抗肿瘤效果表示为%T/C(处理vs.对照),将处理组的肿瘤体积除以对照组并乘以100。
如图8所示,用单独的SIRPAD1-Fc处理引起最小的肿瘤生长抑制,肿瘤生长抑制率仅为93%T/C。而用VEGFR1D2-Fc的处理引起戏剧性的肿瘤生长抑制(32%T/C),向VEGFR1D2-Fc处理加入SIRPAD1-Fc并不进一步增强VEGFR1D2-Fc介导的肿瘤生长抑制(33%T/C)。然而,给人以希望的是,使用重组双功能蛋白MAC-01的处理引起戏剧性的肿瘤生长抑制(25%T/C),这在与用单独VEGFR1D2-Fc或单个分子组合处理的组相比时,显著地为更优(P=0.05)。统计分析的结果示于表2。
表2.在A549s.c.异种移植模型中的肿瘤生长抑制
以上结果显示出,重组双功能蛋白MAC-01具有戏剧性的体内抗肿瘤活性,很可能是通过同时阻断VEGF和SIRP。
尽管本发明已经在以上连同一个或多个实施方式进行了描述,应当理解的是,本发明不限于那些实施方式,且说明意在涵盖可能包括在所附权利要求的宗旨和范围内的所有可变方式、修改方式和等同方式。所有在本文中引用的文献通过引用的方式进一步全部并入本文。
参考文献
1.Chao MP,Weissman IL,and Majeti R.The CD47-SIRPαPathway in Cancer Immune Evasion and Potential Therapeutic Implications.Curr Opin Immunol.2012,24:225-232.
2.Chao MP,Tang C,Pachynski RK,Chin R,Majeti R,Weissman IL.Extranodal dissemination of non-hodgkin lymphoma requires cd47 and is inhibited by anti-cd47 antibody therapy.Blood.2011,118:4890-4901.
3.Chao MP,Alizadeh AA,Tang C,Myklebust JH,Varghese B,Gill S,Jan M,Cha AC,Chan CK,Tan BT,Park CY,et al.Anti-cd47 antibody synergizes with rituximab to promote phagocytosis and eradicate non-hodgkin lymphoma.Cell.2010,142:699-713.
4.Majeti R,Chao MP,Alizadeh AA,Pang WW,Jaiswal S,Gibbs KD,Jr,van Rooijen N,Weissman IL.CD47 is an adverse prognostic factor and therapeutic antibody target on human acute myeloid leukemia stem cells.Cell.2009,138:286-299.
5.Chao MP,Alizadeh AA,Tang C,Jan M,Weissman-Tsukamoto R,Zhao F,Park CY,Weissman IL,Majeti R.Therapeutic antibody targeting of cd47 eliminates human acute lymphoblastic leukemia.Cancer Res.2011,71:1374-1384.
6.Collins SJ.The HL60 promyelocytic leukemia cell line:proliferation,differentiation,and cellular oncogene expression.Blood.1987,70:1233-1244.
7.Xu Y and Scheinberg DA.Elimination of human leukemia by monoclonal antibodies in an athymic nude mouse leukemia model.Clin Cancer Res.1995,1:1179-1187.
8.Lin JJ,Hsu HY,et al.Molecular evidence of anti-leukemia activity of gypenosides on human myeloid leukemia HL-60 cells in vitro and in vivo using a HL-60 cells murine xenograft model.Phytomedicine.2011,18:1075-1085
9.Sun Y,Xu HJ.,et al.Crocin Exhibits Antitumor Effects on Human Leukemia HL-60 Cells In Vitro and In Vivo.Evidence-Based Complementary and Alternative Medicine.2013,2013:1-7.
10.Thomas LH,Friedland JS,Sharland M,Becker S.Respiratory Syncytial Virus-Induced RANTES Production from Human Bronchial Epithelial Cells Is Dependent on Nuclear Factor-κB Nuclear Binding and Is Inhibited by Adenovirus-Mediated Expression of Inhibitor of κBα.Journal of Immunology.1998,161:1007-16.
11.Lin Y,Zhang M,Barnes PF.Chemokine production by a human alveolar epithelial cell line in response to Mycobacterium tuberculosis.Infection and Immunity.1998,66:1121-6.
12.Xu X and Prestwich GD.Inhibition of Tumor Growth and Angiogenesis by a Lysophosphatidic Acid Antagonist in a Engineered Three-dimensional Lung Cancer Xenograft Model.Cancer.2010,116:1739-1750.
13.Coxon A,Ziegler B,et al.Antitumor activity of motesanib alone and in combination with cisplatin or docetaxel in multiple human non-small-cell lung cancer xenograft models.Mol Cancer.2012,11:70.
14.Naumov GN,Nilsson MB,et al.Combined Vascular Endothelial Growth Factor Receptor and Epidermal Growth Factor Receptor(EGFR)Blockade Inhibits Tumor Growth in Xenograft Models of EGFR Inhibitor Resistance.Clin Cancer Res.2009,15:3484-3494.
15.Magda D,Lecane P,et al.mtDNA depletion confers specific gene expression profiles in human cells grown in culture and in xenograft.BMC Genomics.2008,9:521.
- 还没有人留言评论。精彩留言会获得点赞!