使用钴基全接触催化剂生产伯胺的方法与流程
在根据现有技术将腈催化氢化成伯胺中,仲和叔胺的不希望形成可以通过加入氨或碱金属氢氧化物而显著抑制。这里所用催化剂通常包含铁、钴和/或镍作为主组分。例如,己二腈—就量而言为重要的腈—可以在高压条件下在元素铁和氨存在下或者在温和压力下在阮内镍和氢氧化钠溶液存在下氢化而得到六亚甲基二胺。氨的除去和再循环构成资金和能量成本的并非微不足道的成本因素。这对于将腈中间体如异佛尔酮腈、乙腈和二甲氨基丙腈(DMAPN)氢化成相应的伯胺也是如此。
EP-A 913 388描述了一种将腈氢化成伯胺,尤其是将二甲氨基丙腈(DMAPN)氢化成二甲氨基丙基胺(DMAPA)的改进方法。氢化在悬浮钴海绵催化剂如阮内钴、锂化合物和水存在下进行而不加入氨。在氢化之前,将阮内钴催化剂用0.1-100mmol氢氧化锂/g钴海绵催化剂处理。EP-A 913 388的程序的缺点是将悬浮催化剂从氢化输出物除去并将其再循环到氢化中通常具有困难。EP-A 913 388中所述阮内钴催化剂不是未负载的钴催化剂。由于这些阮内催化剂的特殊制备模式,它们具有非常高的表面积。
WO 2003/070 688描述了在镍或钴催化剂存在下将腈氢化成伯胺,所述催化剂已经用碱金属碳酸盐或碱金属碳酸氢盐,优选碳酸钾非现场预处理。“非现场”应理解为是指该催化剂已经在外面且尤其是在腈氢化反应得到胺之前改性。所述合适的催化剂尤其是阮内镍催化剂;任选还可以使用阮内钴催化剂。然而,没有任何实际上在氢化过程中用碱金属化合物连续或重复预处理的描述。此外,没有锂化合物也适合该目的的启示。
WO 2007/104 663 A1也公开了包含锂的固定床钴催化剂在没有氨存在下将腈氢化成伯胺中的用途。这类催化剂可以通过在溶剂如四氢呋喃中用氢气还原式LiCoO2的催化剂前体而制备。这类催化剂前体也可以通过煅烧锂盐和钴盐,例如碳酸锂和碳酸钴的混合物而得到。然而,WO 2007/104 663没有公开可以在氢化过程中重复或连续加入化合物(I)如氢氧化锂。
在还原之后包含至多10重量%锂且描述于WO 2007/104 663中的钴催化剂的其他缺点是存在于意欲氢化的腈且因此存在于氢化混合物中的水导致催化剂失活。另一缺点是当将LiCoO2用作催化剂前体时,至多10重量%锂在该催化剂内键合而不会到达催化活性区以变得在其中呈催化活性。
本发明的目的是要提供使用钴催化剂由相应腈制备伯胺的新方法。
该目的通过一种制备伯胺的方法实现,该方法包括在设备(V1)中在未负载的钴催化剂存在下氢化至少一种腈而得到至少一种伯胺,其中将至少一种化合物(I)重复或连续加入设备(V1)中,其中所述化合物(I)包含至少一种选自碱金属、碱土金属和稀土金属的组分。
通过本发明方法,可以以有利方式制备伯胺,因为使用未负载的钴催化剂,优选呈固定床催化剂形式。这也使得可以在液相中用氢气低压氢化相应的腈。本发明方法的额外特征在于所用未负载的钴催化剂的高活性和使用寿命。由于连续或至少连续加入至少一种化合物(I),尤其是以含水氢氧化锂的形式,额外发现所形成的伯胺具有高产率和选择性。本发明方法的其他优点是该方法还可以在水存在下进行,尤其使用含水的腈,并且无氨操作模式(氢化)也是可能的,不会导致腈转化率和伯胺的选择性显著下降。
在将腈氢化成相应的胺中的共同要求是要基于所用腈实现高转化率,因为未转化的或仅部分转化的腈仅可困难地除去并且可能在随后应用中导致不希望的性能,如臭味和变色。
因此,本发明的优点是本发明方法能够在高选择性和产率下在长反应时间内高转化率地氢化腈。此外,降低了不希望的副产物如仲胺的形成。
这使得可以在更温和的反应条件下,尤其是在更低压力和/或更低温度下进行氢化。
因此,本发明使得经济可行的氢化方法成为可能。更具体而言,降低了例如可能通过未转化的胺与部分氢化的腈(亚胺中间体)反应而出现的仲和叔胺的形成。
更具体而言,本发明方法使得可以高选择性和产率地制备异佛尔酮二胺。更具体而言,可以降低不希望的异佛尔酮腈胺(IPNA)的含量。IPNA例如可以通过异佛尔酮腈与氨的反应形成,它们首先反应得到异佛尔酮腈亚胺,后者随后优先与氢气反应得到异佛尔酮腈胺。
本发明方法同样优选用于制备3-(二甲氨基)丙基胺(DMAPA)。更具体而言,本发明方法可以降低二-DMAPA的含量。它为例如用于生产表面活性物质、肥皂、化妆品、洗发剂、卫生产品、洗涤组合物和作物保护组合物的中间体。DMAPA也用于水处理且用作PU和环氧树脂的聚合催化剂。
还认为本发明方法相对于在悬浮模式下基于阮内钴催化剂的现有技术方法的优点是该类阮内钴催化剂基本不适合以固定床模式使用。这些阮内钴催化剂的安装和拆卸应在惰性条件下进行。
本发明方法也优于基于使用所谓整体式催化剂的现有技术方法。与使用未负载的钴催化剂的本发明方法相比,使用整体式催化剂(其中钴施用于整体式成型体上)就生产而言显著更昂贵;将这些催化剂安装到常规反应器中同样更困难。因此,使用整体式催化剂与本发明方法相比不太经济。
本发明方法也优于基于使用LiCoO2催化剂的方法,因为在LiCoO2催化剂的情况下随着操作时间增加观察到活性丧失,这是因为锂从相应催化剂中浸出。因此,这类催化剂并不非常适合在水存在下进行腈氢化(浸出效应)。
本发明使用未负载的钴催化剂制备伯胺的方法在下文详细定义。
在本发明方法中,原则上可以使用本领域熟练技术人员已知的任何腈。根据本发明,可以使用单一腈或两种或更多种腈的混合物。所用腈(反应物)可以具有一个或者不止一个腈官能团。根据本发明,氢化将所用腈中的腈官能团转化成相应的胺官能团。按照本发明,优选相应的伯胺由每一所用腈制备,在相应腈的其他组分(官能基团)中没有任何化学变化。
优选该腈为脂族单-、二-或三腈,脂环族单-或二腈,α-、β-或ω-氨基腈或烷氧基腈。
进一步优选在本发明方法中使用具有1-30个,尤其是2-18个或2-8个碳原子的脂族单-、二-和/或三腈(线性或支化)或具有6-20,尤其是6-12个碳原子的脂环族单-和二腈或具有1-30个,尤其是2-8个碳原子的α-、β-或ω-氨基腈或烷氧基腈。
此外,优选可以使用具有6-18个碳原子的芳族腈。上述单-、二-或三腈可以被单-或多取代。
特别优选的单腈是用于制备乙胺的乙腈,用于制备丙胺的丙腈,用于制备丁胺的丁腈,用于制备月桂胺的月桂腈,用于制备硬脂基胺的硬脂腈,用于制备N,N-二甲氨基丙基胺(DMAPA)的N,N-二甲氨基丙腈(DMAPN)和用于制备苄胺的苯甲腈。
特别优选的二腈是用于制备六亚甲基二胺(HMD)和/或6-氨基剂己腈(ACN)的己二腈(ADN),用于制备2-甲基戊二胺的2-甲基戊二腈,用于制备1,4-丁二胺的丁二腈和用于制备八亚甲基二胺的辛二腈。
特别优选的环状腈是用于制备异佛尔酮二胺的异佛尔酮腈亚胺(IPNI)和/或异佛尔酮腈(IPN)以及用于制备间苯二甲基二胺的间苯二腈。
特别优选的β-氨基腈是用于制备1,3-二氨基丙烷的氨基丙腈或烷基胺、烷基二胺或链烷醇胺在丙烯腈上的加成产物。例如,可以将乙二胺和丙烯腈的加成产物转化成相应的二胺。例如,可以将3-[2-氨基乙基]氨基丙腈转化成3-(2-氨基乙基)氨基丙基胺,以及将3,3’-(亚乙基二亚氨基)二丙腈或3-[2-(3-氨基丙基氨基)乙基氨基]丙腈转化成N,N’-二(3-氨基丙基)乙二胺。
特别优选的ω-氨基腈是用于制备六亚甲基二胺和己内酰胺的氨基己腈。
进一步特别优选的腈是所谓的“Strecker腈”,如用于制备二亚乙基三胺的亚氨基二乙腈以及用于制备乙二胺(EDA)和二亚乙基三胺(DETA)的氨基乙腈(AAN)。
优选的三腈是三乙腈胺。
在特别优选的实施方案中,在本发明方法中使用N,N-二甲氨基丙腈(DMAPN)来制备N,N-二甲氨基丙基胺(DMAPA)。
在进一步特别优选的实施方案中,在本发明方法中将异佛尔酮腈亚胺用于制备异佛尔酮二胺并且在进一步特别优选的实施方案中,使用己二腈(ADN)来制备六亚甲基二胺(HMD)或制备6-氨基己腈(6-ACN)和HMD。
用于氢化的腈可以包含0.01-10重量%,优选0.1-8重量%,更优选0.1-5重量%,最优选0.1-3重量%水。该水例如可以源自制备所用腈的方法。
氢化本身,即还原所用腈的腈官能团以得到相应的胺官能团,原则上可以通过本领域熟练技术人员已知的任何方法进行。
用于氢化的还原剂可以是氢气或含氢气体。氢气通常以工业级纯度使用。氢气还可以以含氢气体形式使用,即以与其他惰性气体如氮气、氦气、氖气、氩气或二氧化碳的混合物使用。所用含氢气体例如可以是重整器尾气、炼油厂气等,如果且当这些气体不包含任何所用氢化催化剂的催化剂毒物,例如CO时。然而,在该方法中优选使用纯氢气或基本纯氢气,例如含有大于99重量%氢气,优选大于99.9重量%氢气,更优选大于99.99重量%氢气,尤其大于99.999重量%氢气的氢气。
根据本发明,氢化在设备(V1)中进行,其中除了反应物(腈和还原剂)外存在下面更特别规定的其他组分(未负载的钴催化剂和化合物(1))。
所用设备(V1)原则上可以是任何适合氢化且对本领域熟练技术人员已知的设备。设备(V1)原则上可以仅仅为一个单一设备,但还可以使用串联和/或并联连接的两种或更多种这类设备。
本发明方法优选在作为设备(V1)的反应器中进行。额外优选的是在设备(V1)中,尤其是在反应器中,该未负载的钴催化剂以固定床形式排列。
在优选实施方案中,固定床排列包括实际意义的催化剂床,即优选呈下文所述几何形状或形状的负载或未负载的松散成型体。
为此,将成型体引入反应器中。
为了使成型体保持在反应器中并且不从中跌出,通常使用栅格基底或气体和液体可透过的金属片,成型体位于其上。
反应器入口和出口二者处的成型体可以被惰性材料包围。所用惰性材料通常是与上述催化剂成型体具有类似几何形状但在该反应中具有惰性行为的成型体,例如鲍尔环、惰性材料(例如陶瓷、滑石、铝)的球。
或者,可以将成型体与惰性材料混合并作为混合物引入反应器中。
催化剂床(成型体+任何惰性材料)优选具有0.1-3kg/L,优选1.5-2.5kg/L,尤其优选1.7-2.2kg/L的堆积密度(根据EN ISO 6)。
横跨该床的压力差优选小于1000毫巴/m,优选小于800毫巴/m,更优选小于700毫巴/m。优选横跨该床的压力差为10-1000毫巴/m,优选50-800毫巴/m,更优选100-700毫巴/m,尤其是200-500毫巴/m。
在滴流模式中(液体流动方向从顶部向下),压力差由在催化剂床上方测量的压力和在催化剂床下面测量的压力计算。
在液相模式中(液体流动方向从顶部向上),压力差由在催化剂床下面测量的压力和在催化剂床上方测量的压力计算。
合适的固定床反应器例如描述于文章“固定床反应器”(Ullmann's Encyclopedia of Industrial Chemistry,2000年6月15日在线出版,DOI:10.1002/14356007.b04_199)中。
优选在作为设备(V1)的竖式反应器、壳管式反应器或管式反应器中进行该方法。
特别优选在管式反应器中进行该方法。
反应器可以各自作为单独反应器、串联的单独反应器和/或以两个或更多个平行反应器的形式使用。
该反应的具体反应器构造和进行可以取决于待进行的氢化方法、所要求的反应时间和所用催化剂的性质变化。
优选反应器,尤其是管式反应器的高度与直径之比为1:1-500:1,更优选2:1-100:1,尤其优选5:1-50:1。
反应物(原料、氢气、任何液氨)的流动方向通常是从顶部向下或者从底部向上。
更优选反应物(原料、氢气、任何液氨)的流动方向是从顶部向下通过反应器。
在连续模式中催化剂空速通常为0.01-10kg反应物/L催化剂·小时,优选0.2-5kg反应物/L催化剂·小时,更优选0.2-4kg反应物/L催化剂·小时,最优选0.2-2kg反应物/L催化剂·小时。
根据本发明,空塔速度为5-50kg/(m2s),优选8-25kg/(m2s),更优选10-20kg/(m2s),尤其优选12-18kg/(m2s)。
空塔速度v[kg/(m2s)]定义为其中Q为质量流[kg/s]且A为空反应器的横截面积[m2]。
质量流Q又定义为所有供应的反应物料流和再循环料流的质量总和。供应的氢气、循环气体和任何惰性气体不用来计算质量流,因为氢气、循环气体和惰性气体通常在标准氢化条件下存在于气相中。
为了实现高空塔速度,优选将来自氢化反应器的一部分输出物(部分输出物)作为再循环料流(循环料流)再循环到反应器中。可以将循环料流分开供回反应器中,或者更优选可以将其与供应的反应物混合并与其一起供回反应器中。
循环料流与供应的反应物料流的比例优选为0.5:1-250:1,更优选1:1-200:1,尤其优选2:1-180:1。当没有将氨供入该方法中时,循环料流与供应的反应物料流的比例优选在上述范围的上部区域。相反,若将大量氨供入该方法中,则循环料流与供应的反应物料流的比例优选在上述范围的下部区域。
在本发明的优选实施方案中,由设备(V1)排出的料流(S1)包含伯胺并至少部分返回设备(V1),其中优选首先将化合物(I)加入料流(S1)的返回部分中并将其作为料流(S1)的成分供入设备(V1)中,特别优选将化合物(I)作为水溶液加入料流(S1)的返回部分中。
在进一步优选的实施方案中,当该反应在具有纤细设计的反应器中,尤其是在具有纤细设计的管式反应器中进行时可以实现高空塔速度。
因此,反应器的高度与直径之比如上所述优选为1:1-500:1,更优选2:1-100:1,尤其优选5:1-50:1。
未负载的钴催化剂本身对本领域熟练技术人员是已知的。在本发明方法中原则上可以使用任何已知的未负载钴催化剂。优选使用单一未负载的钴催化剂;任选还可以使用两种或更多种不同的未负载钴催化剂的混合物。根据本发明,优选的是该未负载的钴催化剂作为固定床催化剂用于设备(V1)中。
在本发明上下文中,优选不将阮内钴催化剂、基于整料的钴催化剂或基于LiCoO2前体的钴催化剂认为是未负载的钴催化剂。
本发明方法所用未负载的钴催化剂的特征性特征首先是它们
i)具有高比例的催化活性组合物(活性组分),
ii)具有相对低的表面积和/或
iii)惰性载体材料的比例为0或仅具有相对小比例的惰性载体材料。
换言之,这意味着该未负载的钴催化剂主要由催化活性组合物(活性组分)构成。催化活性组合物尤其为钴,其中除了钴外还可以存在其他元素,尤其是金属作为催化活性组合物。催化活性组合物的钴含量通常为至少55重量%且可以至多为100重量%(相应的催化剂仅含有钴作为催化活性组合物)。催化活性组合物中的钴含量优选为至少80重量%,尤其是至少90重量%。额外优选的是未负载的钴催化剂中催化活性组合物的上限为95重量%。因此,剩余5重量%的催化活性组合物由其他元素,尤其是促进剂如锰、钠和/或磷形成。
此外,按照本发明优选的是该未负载的钴催化剂由至少70重量%,优选至少80重量%,更优选至少90重量%,最优选至少95重量%的催化活性组合物构成。
额外优选的是该未负载的钴催化剂包含组分i)以及任选地,组分ii)和/或iii)作为催化活性组合物:
i)钴(Co),
ii)任选地,至少一种选自铁(Fe)、镍(Ni)、钌(Ru)、铑(Rh)、钯(Pd)、锇(Os)、
铱(Ir)和铂(Pt)的其他元素,和/或
iii)任选地,至少一种选自铬(Cr)、锰(Mn)、钼(Mo)、钛(Ti)、锌(Zn)、锡
(Sn)、碱金属、碱土金属、稀土金属和磷(P)的促进剂。
在本发明的进一步优选实施方案中,未负载的钴催化剂的催化活性组合物由55-98重量%钴,0.2-15重量%磷,0.2-15重量%锰和0.2-15重量%碱金属(在用氢气还原之前作为CoO、H3PO4、MnO2或碱金属氧化物计算)构成。优选通过在第一步中在550-750℃的最终温度下和在第二步中在800-1000℃的最终温度下煅烧该催化剂组合物而制备未负载的钴催化剂。这类催化剂例如由EP-A 0 636 409已知。
此外,按照本发明优选的是该未负载的钴催化剂
i)具有的表面积不大于50m2/g,优选不大于40m2/g,更优选不大于30m2/g,和/或
ii)载体材料的比例(基于该催化剂的总质量)不超过10重量%,优选不大于5重量%,更优选不大于1重量%,其中该未负载的钴催化剂尤其不含载体材料。
若本发明方法中所用未负载的钴催化剂包含载体材料,则它优选为ZrO2。相反,按照本发明,优选避免基于氧化铝的载体材料。
在本发明方法中还可以使用通过还原所谓的催化剂前体而制备的催化剂。
催化剂前体包含含有一种或多种催化活性组分、任何促进剂和任选地载体材料的活性组合物。
催化活性组分(组合物)包含上述金属的含氧化合物,例如金属氧化物、其碳酸盐或氢氧化物,如CoO、NiO、CuO和/或其混合氧化物。
在本身请上下文中,表述“催化活性组分”用于上述含氧金属化合物,但不认为暗指这些含氧化合物本身已经呈催化活性。催化活性组分通常仅在还原之后在本发明的转化之后具有催化活性。
掺杂元素(促进剂)优选以不大于10重量%,例如0.1-10重量%,更优选1-5重量%的量存在,在每种情况下基于特定的催化剂前体。
优选的催化剂包含50-99重量%,优选70-99重量%,更优选90-99重量%钴。
优选催化剂前体如公开于EP-A 0 636 409中且在用氢气还原之前包含作为CoO计算为55-98重量%的Co,作为H3PO4计算为0.2-15重量%的磷,作为MnO2计算为0.2-15重量%的锰和作为M2O(M=碱金属)计算为0.2-5.0重量%的碱金属的氧化物混合物,以及根据DE 34 03 377 A1制备的Co/Mn/P催化剂,或公开于EP-A-0 742 045中且在用氢气还原之前包含作为CoO计算为55-98重量%的Co,作为H3PO4计算为0.2-15重量%的磷,作为MnO2计算为0.2-15重量%的锰和作为M2O(M=碱金属)计算为0.05-5重量%的碱金属的氧化物混合物或者公开于EP-A 963 975中且在用氢气还原之前包含22-45重量%ZrO2,作为CuO计算为1-30重量%的铜的含氧化合物,作为NiO计算为15-50重量%的镍的含氧化合物,其中Ni:Cu摩尔比大于1,作为CoO计算为15-50重量%的钴的含氧化合物,分别作为Al2O3和MnO2计算为0-10重量%的铝和/或锰的含氧化合物且没有钼的含氧化合物的氧化物混合物,例如公开于上述引文第17页中的催化剂A,其组成是作为ZrO2计算为33重量%的Zr,作为NiO计算为28重量%的Ni,作为CuO计算为11重量%的Cu以及作为CoO计算为28重量%的Co,或者公开于EP 445 589 B1中且在还原之前包含65重量%CoO,4.7重量%Mn3O4,10重量%CaO或者69重量%CoO,4.9重量%Mn3O4,5.5重量%NiO,10.4重量%Fe2O3和10.0重量%CaO的氧化物混合物。
合适的成型体是具有任何所需几何形状和形状的成型体。优选的形状是片、环、圆柱体、星形挤出物、线料形、车轮或球,特别优选片、环、圆柱体、线料形、球或星形挤出物。非常特别优选线料形或片。
在球的情况下,球形形状的直径优选为10mm或更小,更优选4mm或更小,甚至更优选3mm或更小,尤其优选2.5mm或更小。
在优选实施方案中,在球的情况下,球形形状的直径优选为0.1-10mm,更优选0.5-4mm,甚至更优选1-3mm,尤其优选1.5-2.5mm。
在线料或圆柱体的情况下,长度:直径之比优选为1:1-20:1,更优选1:1-14:1,甚至更优选1:1-10:1,尤其优选1:2-6:1。
线料或圆柱体的直径优选为10mm或更小,更优选5mm或更小,甚至更优选3mm或更小,尤其优选2.5mm或更小。
在优选实施方案中,线料或圆柱体的直径优选为0.1-10mm,更优选0.5-3mm,甚至更优选1-2.5mm,尤其优选1.5-2.5mm。
在片的情况下,片的高度h优选为10mm或更小,更优选4mm或更小,甚至更优选3mm或更小,尤其优选2.5mm或更小。
在优选实施方案中,片的高度h优选为0.1-10mm,更优选0.5-4mm,甚至更优选1-3mm,尤其优选1.5-2.5mm。片的高度h(或厚度)与片的直径D之比优选为1:1-1:5,更优选1:1-1:2.5,甚至更优选1:1-1:2。
在所有其他几何形状中,本发明方法中的催化剂成型体在每种情况下优选具有的当量直径L=1/a’为2mm或更小,更优选1mm或更小,甚至更优选0.7mm或更小,尤其优选0.5mm或更小,其中a’为单位体积的外表面积(mms2/mm3),其中:
其中Ap为成型体的外表面积(mms2)且Vp为成型体的体积(mm3)。
在优选实施方案中,本发明方法中的催化剂成型体在所有其他几何形状中在每种情况下优选具有0.1-2mm,更优选0.1-0.7mm,甚至更优选0.2-0.5mm,尤其优选0.3-0.4mm的当量直径L=1/a’。
成型体的表面积和体积通过已知的数学公式由成型体的几何尺寸计算。
体积还可以由如下方法计算,其中:
1.测定成型体的内部空隙率(例如经由在室温和1巴的总压下测量吸水率[ml/g cat]),
2.测定浸入液体中时成型体的排液量(例如通过借助氦比重瓶的气体置换),以及
3.形成这两个体积的总和。
表面积还可以通过下列方法理论计算,其中确定成型体周围的外套,其具有的曲率半径为最大5μm(以不通过该外套“渗透”进入孔中而包括内孔表面积)并且与该成型体非常紧密地接触(不与载体交叉)。这可以被看作非常薄的膜放置在成型体周围,然后由内部对其施加真空,从而使该薄膜非常紧密地包封该成型体。
所用成型体优选具有0.1-3kg/L,优选1.5-2.5kg/L,尤其优选1.7-2.2kg/L的堆积密度(根据EN ISO 6)。
在优选实施方案中,催化剂以成型体形式用于本发明方法中,该成型体通过浸渍具有上述几何形状的载体材料而生产或者在浸渍之后成型为具有上述几何形状的成型体。
有用载体材料的实例包括碳,如石墨、炭黑、石墨烯、碳纳米管和/或活性炭,氧化铝(γ、δ、θ、α、κ、χ或其混合物),二氧化硅,二氧化锆,沸石,硅铝酸盐或其混合物。
上述载体材料可以通过常规方法浸渍(A.B.Stiles,Catalyst Manufacture-Laboratory and Commercial Preparations,Marcel Dekker,New York,1983),例如通过在一个或多个浸渍步骤中施用金属盐溶液。有用的金属盐通常包括水溶性金属盐如相应催化活性组分或掺杂元素的硝酸盐、乙酸盐或氯化物,如硝酸钴或氯化钴。然后通常干燥并任选煅烧已浸渍的载体材料。
煅烧通常在300-800℃,优选350-600℃,尤其是450-550℃的温度下进行。
浸渍也可以通过“初湿含浸法”进行,其中根据其吸水容量将载体材料用浸渍溶液润湿至最大饱和。或者,浸渍可以在上清液中进行。
在多步浸渍方法的情况下,合适的是在单独的浸渍步骤之间干燥并任选煅烧。当要使载体材料与金属盐以相对大量接触时,有利的是应使用多步浸渍。
为了将多种金属组分施用于载体材料上,浸渍可以用所有金属盐同时进行,或者以任何所需顺序的单独金属盐依次进行。
优选使用已经具有成型体的上述优选几何形状的载体材料。
然而,还可以使用以粉末或碎片的形式存在的载体材料并对已浸渍的载体材料进行成型。
例如,可以调节已浸渍且干燥或煅烧的载体材料。
调节例如可以通过由研磨将已浸渍的载体材料调节至特定粒度而进行。在研磨之后,可以将已调节且已浸渍的载体材料与成型助剂如石墨或硬脂酸混合并进一步加工而得到成型体。
用于成型的标准方法例如描述于Ullmann’s Encyclopedia,2000年电子版,“催化和催化剂”章节,第28-32页以及Ertl等(Ertl,Weitkamp,Handbook of Heterogeneous Catalysis,VCH Weinheim,1997,第98页及随后各页)中。
用于成型的标准方法例如为挤出,压片,即机械压制,或造粒,即通过圆周和/或旋转运动压实。
成型操作可以得到具有上述几何形状的成型体。
调节或成型之后通常进行热处理。热处理中的温度通常对应于煅烧中的温度。
在优选实施方案中,在本发明方法中使用通过其所有组分共沉淀生产的成型体,其中对如此沉淀的催化剂前体进行成型操作。
为此,在加热和搅拌的同时将相应活性组分、掺杂元素的可溶性化合物和任选地,载体材料的可溶性化合物在液体中与沉淀剂混合,直到沉淀完全。
所用液体通常为水。
活性组分的有用可溶性化合物通常包括上述金属的相应金属盐,如硝酸盐、硫酸盐、乙酸盐或氯化物。
所用载体材料的可溶性化合物通常为Ti、Al、Zr、Si等的水溶性化合物,例如这些元素的水溶性硝酸盐、硫酸盐、乙酸盐或氯化物。
所用掺杂元素的可溶性化合物通常为掺杂元素的水溶性化合物,例如这些元素的水溶性硝酸盐、硫酸盐、乙酸盐或氯化物。
在进一步优选的实施方案中,成型体可以通过沉淀施用进行。
沉淀施用应理解为指这样一种生产方法,其中将微溶性或不溶性载体材料悬浮于液体中,然后加入相应金属氧化物的可溶性化合物如可溶性金属盐,随后通过加入沉淀剂将这些沉淀到悬浮的载体上(例如如EP-A2 1 106 600第4页和A.B.Stiles,Catalyst Manufacture,Marcel Dekker,Inc.,1983,第15页所述)。
有用微溶性或不溶性载体材料的实例包括碳化合物如石墨、炭黑和/或活性碳,氧化铝(γ、δ、θ、α、κ、χ或其混合物),二氧化硅,二氧化锆,沸石,硅铝酸盐或其混合物。
载体材料通常呈粉末或碎片形式。
载体材料悬浮于其中的所用液体通常为水。
有用的可溶性化合物包括活性组分或掺杂元素的上述可溶性化合物。
在沉淀反应中,可溶性化合物通常通过加入沉淀剂作为微溶性或不溶性碱性盐沉淀。
所用沉淀剂优选为碱,尤其是无机碱,如碱金属碱。沉淀剂的实例是碳酸钠、氢氧化钠、碳酸钾或氢氧化钾。
所用沉淀剂还可以是铵盐,例如卤化铵、碳酸铵、氢氧化铵或羧酸铵。
沉淀反应例如可以在20-100℃,特别是30-90℃,尤其是50-70℃的温度下进行。
在沉淀反应中得到的沉淀物通常在化学上是不均匀的且通常包含所用金属的氧化物、水合氧化物、氢氧化物、碳酸盐和/或碳酸氢盐的混合物。对于沉淀物的过滤性,可能证明有利的是将它们陈化,这意味着在沉淀之后将它们单独留置一定时间,任选在热条件下或者使空气通过。
通过这些沉淀方法得到的沉淀物通常通过洗涤、干燥、煅烧和调节它们而加工。
在洗涤之后,通常将沉淀物在80-200℃,优选100-150℃下干燥,然后煅烧。
煅烧通常在300-800℃,优选350-600℃,尤其是450-550℃的温度下进行。
在煅烧之后通常调节通过沉淀反应得到的粉状催化剂前体。
调节例如可以通过由研磨将沉淀催化剂调节至特定粒度而进行。
在研磨之后,可以将通过沉淀反应得到的催化剂前体与成型助剂如石墨或硬脂酸混合并进一步加工而得到成型体。
用于成型的标准方法例如描述于Ullmann’s Encyclopedia,2000年电子版,“催化和催化剂”章节,第28-32页以及Ertl等(Ertl,Weitkamp,Handbook of Heterogeneous Catalysis,VCH Weinheim,1997,第98页及随后各页)中。
用于成型的标准方法例如为挤出,压片,即机械压制,或造粒,即通过圆周和/或旋转运动压实。
成型操作可以得到具有上述几何形状的成型体。
调节或成型之后通常进行热处理。热处理中的温度通常对应于煅烧中的温度。
已经通过浸渍或沉淀生产的成型体包含催化活性组分,后者在煅烧之后通常呈其含氧化合物形式,例如作为其金属氧化物或氢氧化物,如CoO、NiO、CuO和/或其混合氧化物(催化剂前体)。
已经通过浸渍或沉淀如上所述制备的催化剂前体通常在煅烧或调节之后还原。还原通常将催化剂前体转化成其催化活性形式。
催化剂前体的还原可以在升高的温度下在搅动或未搅动还原炉中进行。
所用还原剂通常为氢气或含氢气体。
氢气通常以工业级纯度使用。氢气还可以以含氢气体形式使用,即以与其他惰性气体如氮气、氦气、氖气、氩气或二氧化碳的混合物使用。氢气料流还可以作为循环气体在还原中再循环,任选与新鲜氢气混合并且任选在通过冷凝除去水之后。
催化剂前体优选在其中成型体作为固定床排列的反应器中还原。特别优选在其中进行氢气对腈的随后还原的相同反应器中还原催化剂前体。
此外,催化剂前体可以以流化床在流化床反应器中还原。
通常在50-600℃,尤其是100-500℃,更优选150-450℃的还原温度下还原催化剂前体。
氢气分压通常为1-300巴,尤其是1-200巴,更优选1-100巴,压力值在这里和下文中涉及以绝对值测量的压力。
还原的持续时间优选为1-20小时,更优选5-15小时。
在还原过程中,可以供应溶剂以除去反应形成的水和/或例如以能够更快速加热反应器和/或能够更好地在还原过程中除热。溶剂在这里也可以以超临界形式供应。
所用合适溶剂可以是上述溶剂。优选的溶剂是水或醚如甲基叔丁基醚、乙基叔丁基醚、二烷或四氢呋喃。特别优选水或四氢呋喃。合适的溶剂同样包括合适的混合物。
如此得到的成型体在还原之后可以在惰性条件下处理。成型体优选可以在惰性气体如氮气下或者在惰性液体,例如醇、水或使用该催化剂的特定反应的产物下处理和储存。此时可能需要在开始实际反应之前将该催化剂从惰性液体中分离。
催化剂在惰性物质下的储存使得成型体的处理和储存不复杂且无害。
在还原之后还可以使成型体与含氧气流如空气或空气与氮气的混合物接触。
以此方式得到钝化的成型体。钝化的成型体通常具有保护性氧化物层。该保护性氧化物层简化了该催化剂的处理和储存,从而使得例如钝化成型体在反应器中的安装简化。钝化的成型体优选如上所述通过在与反应物接触之前用氢气或含氢气体处理钝化的催化剂而还原。还原条件通常对应于在还原催化剂前体过程中使用的还原条件。活化通常除去保护性钝化层。更优选在氢化过程中将该至少一种化合物(I)反复或连续加入设备(V1)中。
因此,根据本发明,将至少一种化合物(I)重复或连续加入设备(V1)–即氢化中。化合物(I)包含至少一种选自碱金属、碱土金属和稀土金属的组分。化合物(I)本身对本领域熟练技术人员是已知的。优选将一种化合物(I)重复或连续加入设备(V1)中;任选还可以加入两种或更多种化合物(I)的混合物。此外,可能的是至少一种化合物(I)在开始氢化之前已经存在于设备(V1)中,但优选仅在开始氢化之后将化合物(I)加入本发明方法中。
将至少一种化合物(I)加入设备(V1)中可以如所解释的那样是重复或连续的。同时,化合物(I)可以以液体或固体形式加入。这里还应强调的是化合物(I)无需直接加入设备(V1)中;相反,还可以首先在另一设备中,例如在接触设备(V2)中将化合物(I)加入氢化所涉及的组分中的一种或多种中。由该其他设备将化合物(I)输送到设备(V1)中(将化合物(I)间接加入(V1)中)。化合物(I)由该其他设备向设备(V1)中输送/转移或传递通过本领域熟练技术人员已知的方法进行,例如使用泵进行。更具体而言,化合物(I)如上面已经解释的那样可以加入料流(S1)的返回部分中。
在本发明上下文中,“连续加入”化合物(I)应理解为是指相应的加料在延长的期间内进行,优选在至少50%,更优选至少70%,甚至更优选至少90%,尤其是在整个反应时间内进行。优选以使得适合引入(加入)化合物(I)的设备(V1)(例如星形加料器)在上述期间内操作的方式进行连续加入。
在本发明上下文中,“重复加入”化合物(I)应理解为是指相应的加料在规则或不规则时间间隔下进行。优选相应的加料通过出现进一步如下所述的加料条件而发生,尤其是就化合物(I)在来自设备(V1)的输出物中的浓度而言。单独加料之间的时间间隔为至少1小时,优选至少1天。在本发明上下文中,术语“重复”额外应理解为是指至少2次,例如3、4、5、10或甚至100次单独加料。单独加料的具体次数取决于操作期间。后者理想地倾向于无限长。
换言之,重复加入化合物(I)在本发明上下文中应理解为是指几批化合物(I)的相互限定的加料。单一批次的加料可以花几秒至几分钟;根据情况而定,甚至稍微更长的时期是可行的。根据本发明,单独批次的相应加料之间的时间间隔至少10倍于相应批次的加料持续时间长。根据情况而定,在本发明上下文中还可以将“重复加入”的实施方案与“连续加入”的实施方案组合在一起。
重复或连续加入至少一种化合物(I)的准确剂量下文通过氢氧化锂的加料举例说明。然而,细节加以必要的变更之后也适用于本发明化合物(I)的定义所覆盖的所有其他化合物。
输出物中的锂浓度不应低于某一阈值。若该浓度太低,则这表明锂已经从催化剂中浸出。
输出物中的锂浓度也不应太高,因为否则该催化剂丧失其活性(中毒)。
计量加料还可以连续或重复(周期性地)进行。必要的是输出物/反应混合物中的锂浓度锂不变得太高以致于产生沉积物,但足够高到维持在催化剂上的浓度。
更具体而言,特定要求也适用于计量加料位置:
-计量加入的溶液的锂浓度一定不能太高,因为否则在反应器中出现沉积物/堵塞。
-进料应在反应器中存在高流速的点进行,从而使LiOH与反应混合物良好混合。LiOH在水和大多数有机溶剂中具有低溶解度。
碱金属的有用化合物(I)包括Li化合物、Na化合物、K化合物、Rb化合物和Cs化合物,碱土金属的有用化合物(I)包括Mg化合物、Ca化合物、Sr化合物和Ba化合物,稀土金属的有用化合物(I)包括La化合物、Ce化合物、Pr化合物和Nd化合物。
碱金属的优选化合物(I)是锂、钾和铯,碱土金属的优选化合物(I)是镁和钙,稀土金属的优选化合物(I)是镧和铈。
额外优选的是化合物(I)以氧化物、氢氧化物和/或盐的形式使用和/或化合物(I)以水溶液形式使用。有用的盐例如包括硝酸盐、碳酸盐、碳酸氢盐、磷酸盐和羧酸盐,例如甲酸盐和乙酸盐。优选氢氧化物,在本发明上下文中其不包括在术语“盐”内。
额外优选的是使用锂、钾、铯、镁、钙、镧和/或铈的氧化物、氢氧化物或盐的水溶液。
非常特别优选锂化合物,例如氢氧化锂、碳酸锂、磷酸锂、硝酸锂、甲酸锂、乙酸锂的水溶液。这里优选氢氧化锂,并且因此尤其优选氢氧化锂的水溶液。
用于碱金属、碱土金属和稀土金属化合物的溶剂原则上可以是有机溶剂、水或这些溶剂与水的混合物。优选在下文也进一步作为所用腈的溶剂提到的溶剂。它们必须在氢化条件下稳定且充分溶解碱金属、碱土金属和稀土金属化合物。
因此,优选C1-C4醇、水或这些化合物的混合物。非常特别优选水。
按照本发明额外优选的是:
i)用于氢化的腈包含0.01-10重量%水,和/或
ii)一旦作为副产物形成的仲胺比例大于0.5GC面积%就将化合物(I)加入设备(V1)中,和/或
iii)在0.01-10kg反应物/L催化剂·小时的催化剂空速下经由化合物(I)的加入向设备(V1)中的氢化混合物中加入基于设备(V1)中腈的克原子为0.01-500ppm的碱金属、碱土金属和/或稀土金属,其中优选一旦在设备(V1)中作为副产物形成的仲胺比例大于0.5GC面积%就经由化合物(I)的加入向设备(V1)中的氢化混合物中加入基于设备(V1)中腈的克原子为0.01-500ppm的碱金属、碱土金属和/或稀土金属。
“设备(V1)中腈的克原子”在本发明上下文中应理解为指设备(V1)中以克(g)计的腈总量(总重量)。
因此,按照本发明优选的是在0.01-10kg反应物/L催化剂·小时的催化剂空速下经由化合物(I)的加入将基于设备(V1)中以克计的腈总重量为0.01-500ppm的碱金属、碱土金属和/或稀土金属加入设备(V1)中的氢化混合物中,其中优选一旦在设备(V1)中作为副产物形成的仲胺比例大于0.5GC面积%就经由化合物(I)的加入将基于设备(V1)中以克计的腈总重量为0.01-500ppm的碱金属、碱土金属和/或稀土金属加入设备(V1)中的氢化混合物中。
测定GC面积%的方法本身对本领域熟练技术人员是已知的。优选用下列参数测量GC面积百分数:GC柱:60m CP Volamine/WCOT熔凝硅石0.32;温度程序:50℃–10min–15℃/min–240℃–30min。
根据本发明,氢化可以分批、半连续进行,但优选连续进行。
氢化通常在1-300巴,尤其是5-150巴,优选10-100巴,更优选15-60巴的压力下进行。最优选氢化在小于60巴的压力下作为低压方法进行。
温度通常在25-300℃,尤其是50-200℃,优选70-150℃,更优选80-140℃的范围内。
氢气与所用腈的摩尔比通常为2:1-25:1,优选2:1-10:1。氢气可以作为循环气体再循环到该反应中。
同时,优选选择反应条件以使得所用腈和所加入的液体通常呈液相且仅所用氢气或惰性气体在所述反应条件下呈气相。
在尤其连续操作的情况下,氢化的结果可以以分析手段监控,例如通过气相色谱法。若分析揭示伯胺的选择性随着作为副产物形成仲胺如二-DMAPA而下降,则该下降优选可以通过加入所述溶液而逆转。
一旦形成的仲胺比例大于0.5GC面积%,优选大于1GC面积%,更优选大于2GC面积%,甚至更优选大于5GC面积%,尤其大于10GC面积%,则计量加入化合物(I)。
氢化可以在本体中或者在液体中进行。
合适的溶剂例如为C1-C4醇如甲醇或乙醇,C4-C12二烷基醚如乙醚或叔丁基甲基醚,或者环状C4-C12醚如四氢呋喃或二烷,或者烃类如戊烷、己烷、庚烷、辛烷、环己烷。合适的溶剂还可以是上述液体的混合物。在优选实施方案中,该溶剂为氢化产物。
氢化输出物的后处理优选通过蒸馏进行。这作为底部产物除去加入的碱金属、碱土金属和/或稀土金属化合物。
此外,按照本发明优选的是氢化在不存在氨下进行。当选择无氨操作模式时,氢化中的压力优选<85巴。相反,若氢化在氨存在下进行,则同样可以就腈转化率和伯胺选择性观察到额外改善。此外,氨有助于除去氢化热。
此外,按照本发明优选的是设备(V1)为反应器,其中空塔速度优选为5-50kg质量流/m2反应器横截面积·秒。
下面由实施例说明本发明。
实施例1:
在未负载的钴催化剂存在下将二甲氨基丙腈(DMAPN)氢化成二甲氨基丙基胺(DMAPA),重复供入氢氧化锂水溶液
a)催化剂制备和活化
所用催化剂为由90.4重量%钴、5.1重量%锰、0.3重量%钠和3.1重量%磷构成的未负载钴催化剂(线料直径2mm),其根据EP-A 636409的实施例1生产。将40.2g该催化剂引入氢化反应器中并在供应25L(STP)氢气/小时下在12小时内加热至280℃,在供入25L(STP)氢气/小时的同时将该温度保持12小时并在氢气下冷却。
b)DMAPN的氢化
在高度为1米且直径为0.6cm的不锈钢管式反应器(1.4571内管)中以使用上述催化剂作为固定床催化剂的滴流模式进行氢化。将来自氢化的反应输出物冷却、减压、部分排出并部分再循环到反应器中。
用于氢化的进料为包含二甲胺和水的工业级DMAPN。根据GC分析,水含量为0.3-3.6面积%,二甲胺含量为1.4-2.5面积%(GC柱:60m CP Volamine/WCOT熔凝硅石0.32;温度程序:50℃–10min–15℃/min.–240℃–30min)。
氢化总共进行4000小时,最初149小时没有加入LiOH水溶液,然后的3800小时间歇加入LiOH溶液。表1汇编了加料时间和LiOH的量;表2显示了反应器输出物中的锂浓度。
表1:氢氧化锂的计量加料
1氢氧化锂水溶液加料的持续时间
2氢氧化锂水溶液的氢氧化锂浓度
3氢氧化锂水溶液的量
4千克DMAPN/升催化剂·小时
5氢氧化锂与DMAPN的比例(g/g)
6锂与DMAPN的比例(g/g)
7在特定加料时间内进入反应器中的氢氧化锂的绝对量。
氢化通过将200mL粗DMAPA泵入反应器中开始。
氢化温度为90-110℃;压力对最初700小时为85巴,然后对剩余的运行时间为50巴。
在整个运行时间上的催化剂空速为1-1.1kg DMAPN/L催化剂·小时。空塔速度为41-42kg/m2·小时。为了达到这些值,将一部分氢化输出物在减压之后返回到反应器中。
在2544-3500小时的运行时间之间,使用包含5重量%水的DMAPN。
在氢化时间的最初149小时内没有加入LiOH水溶液,氢化输出物仅含有87-90%DMAPA,11-13%二-DMAPA和0.2-0.3%DMAPN(GC面积%);也见对比例2,没有铵的操作。
在149小时之后开始第一次计量加入LiOH水溶液(计量加料期如表1所述)。在加入LiOH水溶液之后,在试验开始之后200小时往前DMAPN转化率在剩余的3500小时内为至少99.4%。DMAPA产率为99.1-99.5%。出现的唯一副产物是量为0.2-0.9%的二-DMAPA。
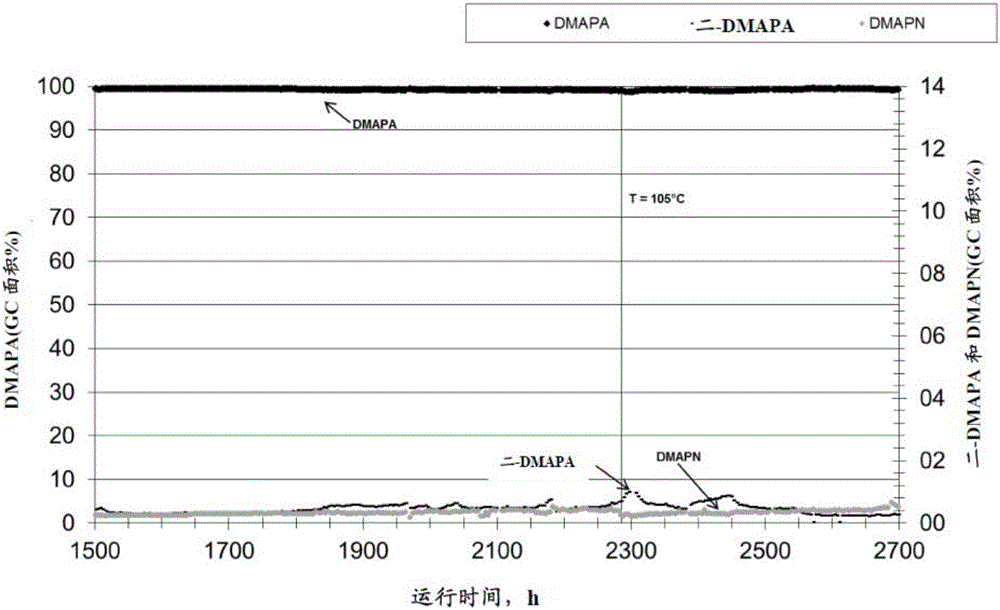
如实施例1所示,在没有计量加入LiOH的操作过程中上升的或高二-DMAPA值通过加入LiOH可能再次降低到更低水平。就此而言,图1-3显示了在实施例1的整个试验持续期间在反应输出物中测量的反应物(DMAPN)、氢氧化锂、产物(DMAPA)和副产物(二-DMAPA)的浓度。
实施例1额外表明少量LiOH可以实现的正面效果,即可以使用高催化剂空速操作并且具有高催化剂使用寿命以及氢化混合物中的水没有干扰性。此外,实现了高DMAPN转化率和DMAPA产率并且可以在没有氨存在下操作。
表2:测定输出物中的锂浓度
对比例2:
在钴催化剂存在下将DMAPN氢化成DMAPA,最初用LiOH水溶液浸渍催化剂一次
如所述将40.2g实施例1中所用催化剂进料活化并将装置中的催化剂用约400g 10%LiOH水溶液从底部向上单程冲洗(1mL/min)。此外,在开始LiOH溶液进料之后约7小时以1000g/L的再循环速率开启液体回路2小时,然后再次关闭LiOH的再循环和计量加料。然后确立所需反应条件(90℃,氢气)并用DMAPA开启该装置。确立814g/h的液体再循环速率和19.4g/h的DMAPN进料速率。将如实施例1所述如此预处理的催化剂用于在90℃和85巴下氢化DMAPN。有规律地测定反应输出物中的锂浓度。在500小时的反应时间之后,氢化输出物由约92%DMAPA,约8%二-DMAPA和小于约0.5%DMAPN构成(GC面积%)。类似于实施例1的图1-3,对比例2的相应值示于图4中。
对比例2表明在呈固定床的未负载钴催化剂情况下,与阮内钴(呈悬浮模式的催化剂;例如见EP-A 913 388的实施例50)相反,催化剂用LiOH的预处理没有引起选择性的任何提高。有规律地测定反应输出物中的锂表明锂持续从装置和催化剂中洗出。该效果在对比例2中可见,尤其是在氢化开始后的大约最初200小时。对于DMAPA的选择性降低和二-DMAPA的增加与从反应器洗出的锂量相关。直到约650小时之后的试验结束,DMAPA的选择性继续持续降低且在反应输出物中仍可检测出锂。
对比例3:
在未负载的钴催化剂存在下和在氨存在下以及在没有氨存在下(没有加入氢氧化锂)将DMAPN氢化成DMAPA
如所述将40.2g实施例1中所用催化剂进料活化。类似于实施例1,首先用200mL粗DMAPA开启该装置。确立90℃的温度和85巴的压力并在该试验的持续过程中维持。连续计量加入16g/h氨和25L(STP)/h氢气;氢化通过进料19.4g/h DMAPN而开始。
在整个运行时间上的催化剂空速为1-1.1kg DMAPN/L催化剂·小时。空塔速度为34-42kg/m2·小时。为了达到这些值,将一部分氢化输出物在减压之后返回到反应器中。在301小时的反应时间之后,氢化输出物由97.8%DMAPA和小于约0.3%DMAPN构成;不存在二-DMAPA(GC面积%)。
在312小时之后,在其他方面不变的试验条件下停止氨进料。实现的DMAPA选择性在不存在氨下低得多。在442小时的反应时间之后,氢化输出物由85.9%DMAPA,小于约0.3%DMAPN和11.1%二-DMAPA构成(GC面积%)。
对比例3表明在没有氨存在下于85巴下研究的未负载钴催化剂上形成大量二-DMAPA并且DMAPA的选择性低。在氨存在下,由于氨的蒸气压,反应压力的明显下降是不可能的。根据对比例3的相应浓度值类似于以前的实施例示于图5中。
- 还没有人留言评论。精彩留言会获得点赞!