通过丙烷部分氧化反应连续制备丙烯酸的方法和设备与流程
本申请要求于2014年10月7日提交的韩国专利申请第10-2014-0134847号以及于2015年10月7日提交的韩国专利申请第10-2015-0141252号的权益,其全部内容通过引用在此并入本申请中。本发明涉及一种通过丙烷的部分氧化连续制备丙烯酸的方法和设备。
背景技术:
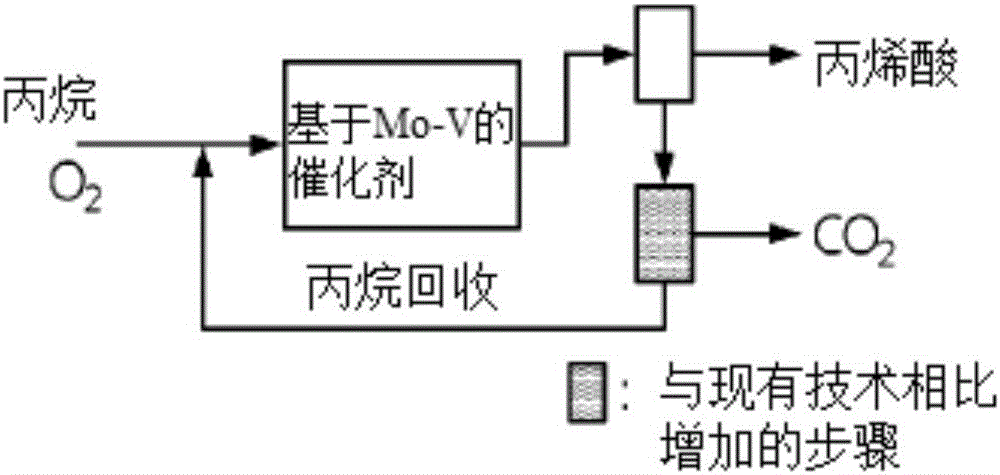
:丙烯酸被用作粘合剂,并且丙烯酸是用于例如制备在水介质中分散而使用的聚合物的单体的重要化学品。主要以首先将丙烯氧化为丙烯醛,然后继续氧化的方式来制备丙烯酸。具体来讲,图1示出由丙烯生产丙烯酸(AA)的工艺。如该图所示,使用基于Mo-Bi的第一步催化剂以及基于Mo-W的第二步催化剂使丙烯与空气中的氧气反应,由此丙烯几乎被全部消耗掉(97%以上的转化率)。在反应产物当中,包含AA的可冷凝的含氧产物在吸收塔中被冷凝为液相,接着被转移至AA分离塔中。大约30%的料流(不仅包括未反应的丙烯还包括氮气(N2)、氧气(O2)、一氧化碳(CO)、二氧化碳(CO2)和水(H2O))被回收。回收的料流包含大量的氮气(N2),大约25体积%至30体积%的水(H2O)以及少量的氧气(O2)、一氧化碳(CO)和二氧化碳(CO2)。回收的料流在反应器的入口处与丙烯和空气混合接着被引入反应器中,并且其剩余的大约70%被清除接着在焚烧炉中被焚烧。供应到反应器的料流与回收的料流一起在反应器的入口处具有以下组成:7%丙烯、12.6%氧气(O2)(来自空气)、72.4%氮气(N2)(47.4%来自空气,25%来自回收的料流),以及8%水(H2O)(来自回收的料流)。因为低的丙烷转化率以及丙烯酸的选择性,由丙烷制备丙烯酸的基于丙烷的丙烯酸生产工艺(POA)尚未被商业化。图2示意性示出基于丙烷的丙烯酸生产工艺,该工艺未作改变地应用基于丙烯的丙烯酸生产工艺。该方法的缺点在于未反应丙烷的过度损失。例如,USP7304014公开了单程68%的丙烷转化率以及80%的丙烯酸选择性,但其问题在于当将该基于丙烷的丙烯酸生产工艺应用到基于丙烯的丙烯酸生产工艺时,32%未反应的丙烷成分中仅有30%被回收,并且其剩余的70%在焚烧炉中被清除,令人遗憾地造成22.4%(0.32*70)的丙烷损失。在基于丙烷的工艺中主要使用的催化剂是MoVTeNb。在基于MoVTeNb的催化剂的存在下,当丙烷/O2比例高时,丙烷转化率低。另一方面,当丙烷/O2比例低时,丙烷转化率提高,但如CO,CO2等的COx的选择性也提高,也就是说,含氧的碳的选择性提高,由此降低了丙烯酸的产率或丙烯酸的选择性。目前可用的基于丙烷的丙烯酸生产工艺的问题在于因为大量未反应的丙烷以及不可冷凝的气体,如CO、CO2或丙烯,令人遗憾地造成在副产物的分离中过多的能量消耗而且需要额外的单元来除去CO和CO2。技术实现要素:技术问题在目前可用的基于丙烷的丙烯酸生产工艺中,使用氮气作为载气。然而,当使用氮气载气时,必须额外地从副产物中分离COx以便回收未反应的丙烷,而且,必须添加氧气来代替空气。如图2所示,在不分离COx的情形中,未反应的丙烷损失很大。而且,为了提高转化率,当丙烷/O2比例降低时,需要大量的能量分离大量产生的CO和CO2。此外,丙烯酸产率明显低。因此,本发明意图提供一种通过丙烷的部分氧化制备丙烯酸的方法,该方法消耗较少的能量并且不贵。此外,本发明意图提供一种制备丙烯酸的方法,其能够防止在用于丙烷的部分氧化的催化剂上的焦炭沉积。另外,本发明意图提供一种连续制备丙烯酸的方法,其能够防止反应器内部温度的过度升高并且防止产生另外的副产物。另外,本发明意图提供一种连续制备丙烯酸的方法,在该方法中可回收利用副产物。技术方案本发明的一个方面提供一种连续制备丙烯酸的方法,其包括以下步骤:(1)使用催化剂,而不使用氮气使包含丙烷、氧气、水蒸气以及二氧化碳的进料经历部分氧化,由此得到含丙烯酸的混合气体;(2)使用吸收溶剂将所述含丙烯酸的混合气体分离成含丙烯酸的溶液以及气体副产物;(3)通过纯化将所分离的含丙烯酸的溶液分离成丙烯酸溶液以及液体副产物;以及(4)不进行额外的分离将所分离的气体副产物回收至所述进料中。在本发明的优选的实施方式中,所述气体副产物可以包括选自丙烷、丙烯、氧气、一氧化碳、二氧化碳以及水蒸气中的至少一种,并且可回收所述回收的气体副产物从而使其适合进料的摩尔比。可保持包含步骤(4)的分离的气体副产物的进料的组成摩尔比以使其适合步骤(1)中的进料的组成摩尔比。在本发明的优选的实施方式中,可在步骤(1)和(2)之间进一步使用包含选自铑(Rh)、铱(Ir)、钯(Pd)和铂(Pt)中的至少一种的催化剂将一氧化碳氧化成二氧化碳。本发明的另一个方面提供一种用于连续制备丙烯酸的设备,其包括:(1)部分氧化反应器,其被构造成通过使用催化剂使经由进料传输线传输的包含丙烷、氧气、水蒸气以及二氧化碳的进料经历部分氧化来制备含丙烯酸的混合气体;(2)吸收塔,其被构造成将所述含丙烯酸的混合气体分离成含丙烯酸的溶液以及气体副产物并被构造成吸收所述含丙烯酸的溶液;(3)蒸馏塔,其被构造成将所述含丙烯酸的溶液分离为丙烯酸溶液;以及(4)回收传输线,其被构造成回收所分离的气体副产物。有益效果根据本发明,可以提供通过丙烷的部分氧化制备丙烯酸的方法,该方法消耗较少的能量并且不贵。此外,本方法能够防止催化剂上的焦炭沉积、反应器内部温度的过度升高以及另外转化成COx以及含氧化合物。此外,根据本发明,可以通过回收来利用除了丙烯酸以外的副产物。具体来讲,在本发明中,当使用二氧化碳(CO2)作为载气时,不需除去二氧化碳(CO2)的另外的分离单元,就可以几乎完全回收未反应的丙烷。因此,由于简单的制备工艺以及显著降低的生产成本,该连续制备丙烯酸的方法是有利的。附图说明图1示出基于丙烯的丙烯酸生产工艺;图2示意性示出示例性的基于丙烷的丙烯酸生产工艺(POA),该工艺未作改变地应用基于丙烯的AA生产工艺;以及图3示意性示出根据本发明的优选实施方式进行丙烯酸制备工艺的设备。具体实施方式下文中,将给出本发明的详细描述。列出以下描述仅用于说明本发明的实施方式,而不将其理解为限制由权利要求限定的范围,即使其包含限制性的表述。在使用基于丙烷的工艺制备丙烯酸的方法中,当使用氮气作为载气时,不仅需要从副产物中额外地分离COx而且需要分离氮气,这是不期望的。而且,由于未反应丙烷的大量损失而导致经济效益失去价值。因此,本发明人已发现这样的事实:通过在丙烷的部分氧化期间使用二氧化碳(CO2)作为载气来解决以上问题,从而完成本发明。因此,本发明的一个方面提出连续制备丙烯酸的方法,其包括以下步骤:(1)使用催化剂使包含丙烷、氧气、水蒸气以及二氧化碳的进料经历部分氧化,由此得到含丙烯酸的混合气体;(2)将所述含丙烯酸的混合气体分离成含丙烯酸的溶液以及气体副产物;(3)从所述含丙烯酸的溶液中分离丙烯酸溶液;以及(4)将所分离的气体副产物回收至所述进料中。在本发明中,术语“含丙烯酸的混合气体”是指可在通过部分氧化制备丙烯酸时产生的混合气体。例如,如下示例出在催化剂的存在下进行的POA工艺,其显示60%的丙烷转化率、80%的AA选择性以及20%的CO2选择性。在吸收塔中将包含AA可冷凝含氧产物冷凝为液体,接着将其传输至AA分离塔,并回收大部分(96%)的包含未反应的丙烷的二氧化碳(CO2)料流,其中仅少量(4%)被清除。也就是说,其中的一部分以与通过POA催化产生的二氧化碳(CO2)的量对应的量被清除和除去。利用了CO2为在丙烯酸吸收单元未被冷凝的主要副产物这一情况。具体来讲,在回收的料流中的水(H2O)的量可为约25体积%至30体积%。因此,在反应器的入口处的进料可由大约7%丙烷、14%O2(分子O2)、50%至59%CO2以及20%至29%H2O组成。如果水的量大于25体积%至30体积%,则从吸收塔排出的二氧化碳(CO2)料流可包含大量的挥发性副产物。另一方面,如果其量小于25体积%,则在吸收时必须使用过量的水,因此在分离步骤中需要大量的能量。以下描述根据本发明的制备丙烯酸的方法。根据本发明的实施方式,制备丙烯酸的方法包括(1)通过部分氧化得到含丙烯酸的混合气体。如上所述,可在催化剂的存在下使包含丙烷、氧气、水蒸气以及二氧化碳的进料经历部分氧化来获得所述含丙烯酸的混合气体。所述部分氧化可以是气态反应,并且可在通常条件下使用本发明所属的
技术领域:
(下文中,称为“本领域”)中通常的部分氧化反应器来进行所述气态氧化反应。在本发明中,丙烷是用于生产丙烯酸的原料,并且氧气用作部分氧化丙烷的氧化剂。在本发明中,术语“部分氧化”是指不是完全氧化而是部分氧化的物质,因此不仅产生期望的氧化物质而且产生未反应的物质以及副产物。在本发明的优选的实施方式中,丙烷与氧气的摩尔比可在1:0.5至3,并且优选1:1至2.5的范围内。如果氧气的摩尔比小于0.5,则用于转化成期望的丙烯酸的氧气的量太少,因此可导致低的转化率和选择性,并且需要用于回收未反应的丙烷的大容量单元。另一方面,如果氧气的摩尔比超过3,则可提高丙烷的转化率但也可提高二氧化碳(CO2)、一氧化碳(CO)、乙酸和丙酸的选择性,令人遗憾地降低丙烯酸的产率。因此,优选将其摩尔比限定在以上范围。同时,在本发明中,如以下反应式所示,丙烷可被氧化成丙烯酸。[反应式1]CH3CH2CH3+2O2→CH2CHCOOH+2H2O在本发明的优选的实施方式中,可在200℃至500℃,优选在300℃至450℃下进行所述部分氧化。如果反应温度低于200℃,则用于活化丙烷的能量低,因此反应不能发生。另一方面,如果反应温度高于500℃,则通过部分氧化产生的丙烯酸可被进一步氧化成一氧化碳(CO)和二氧化碳(CO2),因此选择性降低。因此,优选将反应温度限定在以上范围。在根据本发明的部分氧化中,反应压力为0psig至75psig,优选为5psig至50psig。如果是以上的压力范围,则工艺成本低,反应选择性高。接下来,描述水蒸气。水蒸气的作用是提高丙烯酸的选择性以及改变丙烷的爆炸限度从而提高催化剂的稳定性。基于进料的总摩尔%,包含的水蒸气的量可为5摩尔%至50摩尔%,优选10摩尔%至40摩尔%,并且更优选15摩尔%至30摩尔%。如果水蒸气的量小于5摩尔%,则由于丙烷而形成的焦炭可沉积在催化剂上,从而使催化剂失活。如本领域的技术人员通常所使用的,术语“焦炭”指代还原烃所产生的碳沉积物。另一方面,如果水蒸气的量超过50摩尔%,则沉积在催化剂上的焦炭的量可能降低,但添加到反应器中的丙烷的量也将降低,不期望地造成低的丙烯酸生产率。另外,为了在反应期间使水蒸气的量保持恒定,在反应后应另外分离排出气体。因此,优选将水蒸气的摩尔量限定在以上范围。接下来,描述二氧化碳。在本发明中,二氧化碳是载气,并且被用作具有比氧气更低的氧化力的温和的氧化剂以便氧化沉积在催化剂上的焦炭。在本发明中,所述温和的氧化剂,也即二氧化碳,具有比氧气(O2)或一氧化碳(CO)更低的氧化力,并且具有氧化其他物质并使其自身还原的能力。而且,二氧化碳具有比氮气(N2)和水(H2O)更高的热容,因此在部分氧化期间在反应器内部温度的迁移以及升高中发挥作用。通常,将氮气(N2)用作丙烯酸制备中的载气。为了回收氮气(N2),不得不进一步从副产物中分离氮气并供应空气以补偿缺乏的氮气。因此,该制备设备进一步需要分离单元以及用于供应纯氮气或作为氮源的空气的压缩机以补偿缺乏的氮气。当载气为氮气(N2)时,用于丙烷的部分氧化的催化剂是酸性的,因此可能沉积焦炭,不期望地使催化剂失活。然而,在本发明中,使用二氧化碳作为载气,因此省掉以上额外的步骤和/或单元。因此,无需额外的步骤和/或单元,即降低工艺成本,由此降低制备成本。当使用具有氧化力的二氧化碳作为载气时,在催化剂上的焦炭被氧化成COx,使得可以防止催化剂上的焦炭沉积。另外,在部分氧化的副产物中包含的二氧化碳被再次利用,由此省掉了在丙烯酸的连续制备期间用于连续供应二氧化碳进料的步骤和/或单元。因此,可以降低工艺成本并简化工艺,从而提高处理效率。由于利用氧气的丙烷的部分氧化是放热的,为了防止温度过度升高不得不除去产生的热。此时,载气和/或水蒸汽可用于吸收该热,氮气(N2)在400℃下具有22.1kJ/kmol.K的热容而水蒸气在400℃下具有37.1kJ/kmol.K的热容。相比之下,二氧化碳(CO2)在400℃下具有48.9kJ/kmol.K的热容,因此在200℃至500℃的部分反应温度下其热容高于氮气(N2)和水蒸气的热容。由此,防止了温度过度升高而且可以防止另外转化成COx以及含氧化合物。在本发明的优选的实施方式中,基于进料的总摩尔%,二氧化碳的包含量可为40摩尔%至80摩尔%,优选45摩尔%至75摩尔%,并且更优选40摩尔%至70摩尔%。如果二氧化碳的量低于40摩尔%,对于抑制反应器内部温度升高该范围很低,所以需要额外的冷却剂来降低反应器的内部温度。另一方面,如果二氧化碳的量超过80摩尔%,则用于制备丙烯酸的丙烷的量降低,不期望地降低每体积反应器中丙烯酸的生产率。因此,优选将二氧化碳的摩尔量限定在以上范围。下文描述催化剂。用于本发明的部分氧化的催化剂可包括在领域中通常有用的催化剂,并且优选使用由以下化学式1表示的催化剂。然而,本发明不限于此。在本发明中,可使用由以下化学式1表示的催化剂。使用以下催化剂的原因是虽然高温下碲(Te)蒸发,仍可显示最高的丙烯酸产率。[化学式1]MoVaTebNbcOx在化学式1中,a、b、c和x为相对于1份Mo的比例。在化学式1中,a为0.006至1,并且优选0.09至0.8的数。b为0.006至1,并且优选0.04至0.6的数,并且c为0.006至1,并且优选0.01至0.4的数。并且,x为连接到其他元素的氧的量,并取决于其他元素的氧化态。在化学式1中,Mo是钼,V是钒,Te是碲,Nb是铌,O是氧。化学式1的催化剂在本发明的部分氧化期间经历低还原,并通过与气态氧反应被再度氧化。其活性稍降低,但在使用预定的时间段后,在氧气、水蒸气以及二氧化碳的存在下通过加热容易再生。再生后,可以恢复催化剂原来的活性,并可将该催化剂用于其他反应循环。在本发明的实施方式中,为了防止反应器温度急剧升高可将催化剂放置在多管反应器中。根据本发明的化学式1的催化剂制备如下。在35℃下,将0.234g偏钒酸铵、0.352g碲酸和1.178g仲钼酸铵添加到50ml蒸馏水中得到澄清溶液。将预溶解在8ml蒸馏水中的0.348g草酸铌铵添加到该溶液中然后混合。使得到的混合物搅拌下混合3小时,接着加入硫酸。使用旋转蒸发仪使由此制备的浆液中的溶剂蒸发,并在120℃的烘箱中使所述浆液完全干燥,研磨制粒,筛分为180μm至250μm,并在空气气氛200℃下烧制2小时以及在氮气气氛600℃下烧制2小时。该方法是示意性地由浆液制备催化剂,或可选地,可通过在高压釜中水热处理、过滤、干燥以及烧制来制备所述催化剂。接下来,描述步骤(2)。通过步骤(1)中的部分氧化产生的含丙烯酸的混合气体不仅可包括目标产物,也就是丙烯酸,而且可以包括有机和/或无机副产物。有机副产物的实例可包括丙烯醛、丙酸、丙酮、乙酸、高沸点副产物、未反应的丙烷和/或丙烯,而无机副产物的实例可包括一氧化碳、二氧化碳以及水蒸气。在本发明的步骤(2)中,将吸收溶剂添加到步骤(1)中产生的含丙烯酸的混合气体中,从而可将其分离为含丙烯酸的溶液和气体副产物。可以没有限制地进行使用所述吸收溶剂的分离步骤,只要其通常是有用的。可以没有限制地使用所述吸收溶剂,只要其通常是有用的,并且所述吸收溶剂优选为水。在本发明的步骤(2)中,所述气体副产物的沸点低于吸收溶剂的沸点而且在使用的吸收溶剂中溶解性低,因此留在气相中并可被分离。在含丙烯酸的溶液中包含的副产物的沸点高于步骤(2)中吸收溶剂的沸点并且低于部分氧化反应温度。在含丙烯酸的溶液中包含的副产物在部分氧化的温度下存在于气相中,接着在与吸收溶剂接触时从可冷凝的气体转化成液相,从而被分离。在本发明的优选的实施方式中,所分离的气体副产物可包括未反应的丙烷、副产物的丙烯、一氧化碳、二氧化碳和水蒸汽。所述含丙烯酸的溶液指代不包括液相中的气体副产物而包括含丙烯酸的混合气体的溶液。所述含丙烯酸的溶液可包括丙烯酸、丙烯醛、丙酸、丙酮、乙酸和/或高沸点副产物。所述气体副产物可包括气相中痕量或少量的含丙烯酸的溶液,而所述含丙烯酸的溶液可包括痕量或少量的气体副产物。这种状态可根据吸收条件而变化,但优选的是气体副产物和含AA的溶液完全分离。为了提高效率,在步骤(2)中排出的含丙烯酸的溶液包括40重量%至90重量%,优选50重量%至90重量%,并且更优选50重量%至80重量%的丙烯酸。在本发明的步骤(2)中,将水添加到含丙烯酸的混合气体中。接着将其分离为包括具有比水的沸点低的沸点并且在水中溶解性低的副产物的气体副产物,并且还分离为包含具有比水的沸点高的沸点的副产物的含丙烯酸的溶液。可按照如上所述来进行本发明的步骤(2),但本发明不限于此。在本发明的优选的实施方式中,所述气体副产物可包括选自未反应的丙烷、副产物的丙烯、一氧化碳、二氧化碳和水蒸汽中的至少一种。由于所述气体副产物的组成与进料的组成近似,既无需将其完全除去也无需额外分离,可通过回收对其适当利用。在所述气体副产物中,烯烃,如丙烯以非常小的量存在,并且在催化剂的存在下可被转化为丙烯酸,并且不会显著地影响反应,因此不将其从其他的副产物中分离。因此,在本发明的工艺中,从如丙烷的烷烃中分离烯烃没有必要也不优选。所述含丙烯酸的混合气体和/或所述气体副产物可包括一氧化碳。一氧化碳可以以与二氧化碳的量近似相等的量产生。或者,在本发明中通过使用基于MoVaTebNbcOx的催化剂部分氧化丙烷所产生的副产物中,可产生比二氧化碳的量大的量的一氧化碳。因此,在连续进行根据本发明的制备丙烯酸的方法的情形中,产生的一氧化碳(CO)的量大于产生的二氧化碳(CO2)的量,因此一氧化碳(CO)随着反应时间的延长在反应器中累积,使得可以进行正常的操作。危险的是,除非在POA催化剂的存在下通过与氧气反应将一氧化碳(CO)转化成二氧化碳(CO2),否则与产生的一氧化碳(CO)的浓度相比,稳定状态下一氧化碳(CO)的浓度增加约24倍,因此进料的组成将下降到爆炸范围内。因此,优选将一氧化碳氧化成二氧化碳。在步骤(2)之前以及步骤(1)之后,或从步骤(4)循环回步骤(1)期间,优选地进一步将一氧化碳氧化成二氧化碳。这是因为在进料的部分氧化期间未耗尽的未反应的氧气与金属催化剂反应从而将一氧化碳转化成二氧化碳。在本发明的优选的实施方式中,在步骤(1)和步骤(2)之间,可进一步使用包括选自铑(Rh)、铱(Ir)、钯(Pd)和铂(Pt)中的至少一种的催化剂将一氧化碳氧化成二氧化碳。而且,在从步骤(4)循环回步骤(1)期间,可进一步使用包括选自铑(Rh)、铱(Ir)、钯(Pd)和铂(Pt)中的至少一种的催化剂将一氧化碳氧化成二氧化碳。接下来,描述步骤(3)。在工艺期间,为了防止有机副产物的累积,可以对在步骤(2)中分离的含丙烯酸的溶液中包含的有机副产物进行分离。因此,在本发明的步骤(3)中,对含丙烯酸的溶液纯化从而将其分离为丙烯酸溶液和液体副产物。可以没有限制地进行在本发明的步骤(3)中的纯化,只要其通常是有用的。优选使用利用沸点之间差异的蒸馏法,但本发明不限于此。在步骤(3)中,将步骤(2)中分离的含丙烯酸的溶液冷却以初步除去高沸点副产物,接着使用热和共沸溶剂通过蒸馏将其分离成丙烯酸溶液和液体副产物。所述液体副产物可包括丙烯醛、丙酸、丙酮、乙酸和/或高沸点副产物,而所述丙烯酸溶液可以是通过除去所述液体副产物而仅包含丙烯酸的溶液。所述丙烯酸溶液可包含痕量或少量的液体副产物,但优选地仅包含丙烯酸。可使分离的丙烯酸溶液经历萃取蒸馏和/或结晶,由此得到丙烯酸,并且可以除去液体副产物。在本发明的优选的实施方式中,回收分离的丙烯酸溶液从而得到丙烯酸,并且分离并除去所述液体副产物,并且可以通过回收来利用所述气体副产物。接下来,描述步骤(4)。在步骤(2)中分离的气体副产物可包括选自丙烷、丙烯、氧气、一氧化碳、二氧化碳和水蒸汽中的至少一种,并且可以回收所述分离的气体副产物。所述分离的气体副产物的组成与进料的组成近似,并且可通过回收被适当地利用。不需要任何另外的纯化或分离,可将所述气体副产物直接回收至步骤(1)的进料中。在回收到进料中时,可以被包含在进料中的状态回收所述气体副产物,或者可通过另一个料流进行回收,由此使其与进料经历部分氧化。在本发明的优选的实施方式中,可以回收包括由步骤(4)回收的气体副产物的进料使其适合步骤(1)的进料的组成摩尔比例。如上所述,进料的摩尔比如下:丙烷与氧气的摩尔比为1:0.5至3,基于进料的总摩尔%,水蒸气的量为5摩尔%至50摩尔%,而二氧化碳的量为40摩尔%至80摩尔%。使包含步骤(1)的进料与步骤(4)的回收气体副产物的进料的组成摩尔比例保持恒定从而可以连续制备丙烯酸。下文详细描述包含步骤(4)中回收的气体副产物的进料。在步骤(4)中回收步骤(2)中分离的气体副产物,并且以被包含在流向步骤(1)的进料中的状态供应所述回收的气体副产物。步骤(4)的回收的气体副产物是通过步骤(1)中新鲜进料的部分氧化产生或留下来的副产物。具体来讲,在本发明中,所述回收的气体副产物可包括在通过步骤(1)中新鲜进料的部分氧化部分消耗之后残留的丙烷、氧气、水蒸汽和二氧化碳,并且可包括通过部分氧化而另外产生的水蒸汽和二氧化碳。因此,可通过除去部分水蒸气和二氧化碳以及通过仅添加丙烷和氧气使其适合进料的摩尔比来将所述回收的气体副产物供应到步骤(1)的进料中。必要时,可向步骤(1)仅供应包括丙烷和氧气(在回收气体副产物中不足)的所需进料。可以减少包含所述回收气体副产物的进料的供料量,而不是新鲜进料的供料量。因此,本发明省去了在相关领域中使用的用于分离二氧化碳的额外的单元和步骤。而且,可以减少为连续制备丙烯酸而供应的进料量。因此,本发明能够通过温度控制、清除痕量的气体以及供应少量的进料来稳定连续地制备丙烯酸。由此,本发明有效地降低成本和简化工艺。而且,可以调节进料的摩尔比以使其适合期望的丙烯酸选择性以及丙烷转化率。同时,常规的基于丙烷的丙烯酸制备方法显示低于50%的丙烷转化率以及非常低的丙烯酸产率。然而,除了载气以外,在与常规方法相同的条件下,根据本发明的丙烯酸的制备方法显示40%以上的丙烷转化率以及70%以上的丙烯酸选择性。使用以下等式1和2计算丙烷转化率以及所述选择性。[等式1]丙烷转化率(%)=(反应的丙烷摩尔数/供应的丙烷摩尔数)×100[等式2]丙烯酸选择性(%)=(产生的丙烯酸摩尔数/反应的丙烷摩尔数)×100虽然在相关技术中仅意图提高转化率,而在本发明中,即使在与相关技术中相同的条件下在近似的丙烷转化率时仍可保持高的选择性。另外,可以回收进料由此保证高的丙烯酸产率。此外,可回收部分副产物,从而降低成本。本发明的另一方面提出用于连续制备丙烯酸的设备,其包括:(1)部分氧化反应器,其被构造成通过使用催化剂使经由进料传输线传输的包含丙烷、氧气、水蒸气以及二氧化碳的进料经历部分氧化来制备含丙烯酸的混合气体;(2)吸收塔,其被构造成将所制备的含丙烯酸的混合气体分离成含丙烯酸的溶液以及气体副产物并被构造成吸收所述含丙烯酸的溶液;(3)蒸馏塔,其被构造成将所述含丙烯酸的溶液分离为丙烯酸溶液;以及(4)回收传输线,其被构造成回收所分离的气体副产物。可将所述回收传输线连接到所述进料传输线。或者,可将所述回收传输线连接到所述部分氧化反应器。在本发明的优选的实施方式中,可进一步设置含有包括选自铑(Rh)、铱(Ir)、钯(Pd)和铂(Pt)中的至少一种的催化剂的氧化单元。在本发明的另一个优选的实施方式中,可进一步设置控制单元,其被构造成控制所述回收气体副产物以便适合进料的组成摩尔比例。所述控制单元可以设置于回收传输线。用于连续制备丙烯酸的设备可以使用根据本发明的连续制备丙烯酸的方法。因此,关于丙烷、氧气、水蒸气和二氧化碳的量、气体副产物、催化剂、部分氧化温度以及吸收溶剂仍与上述相同。可使用本领域中常用的静态设备、动态设备或它们的组合来执行根据本发明的通过丙烷的部分氧化制备丙烯酸的方法。例如,可在包括部分氧化反应器、氧化单元、吸收塔、控制单元、蒸馏塔和传输线,并且还包括有机副产物吸收塔和/或冷却塔的制备设备中进行所述方法。然而,本发明不限于此。图3示出用于制备丙烯酸的设备,其包括丙烷的部分氧化反应器,同时根据本发明的实施方式回收气体副产物。所述设备包括部分氧化反应器11、氧化单元12、吸收塔14、控制单元16以及蒸馏塔20,还包括用于连接各个单元的传输线10、13、15、17、18、19、21、22、23。具体来讲,包含丙烷、氧气、水蒸汽和二氧化碳的进料借助进料传输线10在包含催化剂的部分氧化反应器11中经历部分氧化,由此制备含丙烯酸的混合气体。在所制备的含丙烯酸的混合气体通过含有Pt催化剂层的氧化单元12时,一氧化碳被氧化成二氧化碳。接着,含丙烯酸的混合气体通过从其顶部喷水的吸收塔14,从而被分离为含丙烯酸的溶液以及气体副产物。所述含丙烯酸的溶液经由传输线19被传输至蒸馏塔20,从而被分离成丙烯酸溶液和液体副产物。使所分离的丙烯酸溶液经历蒸馏和重结晶,从而得到丙烯酸。使用回收传输线18的控制单元16处理在吸收塔中分离的气体副产物。由此将二氧化碳和水蒸气的量调节到与新鲜进料的摩尔%相适应,从而将气体副产物部分分开并回收。为了使其与新鲜进料的摩尔%相适应,添加丙烷和氧气并与所述气体副产物合并,将得到的料流经由传输线18、10供应到部分氧化反应器11中。所述回收的气体副产物包括丙烯、未反应的丙烷、二氧化碳和水蒸汽,并且可调节所述回收的气体副产物与另外供应的丙烷和氧气的总摩尔比以使其一致。在本发明的优选的实施方式中,可在部分氧化反应器中设置氧化单元或可将其布置在部分氧化反应器与吸收塔之间。为了提高含丙烯酸的混合气体与吸收溶剂的接触效率,可将吸收塔14设置为具有如拉西环、鲍尔环、鞍形物、纱布或结构填充的填充物的填充柱,或常见的多级柱。关于蒸馏塔20,例如,蒸馏塔20a可为用于分离醛或烯烃的低沸点分离塔,并且蒸馏塔20b经由传输线22可用于分离乙酸,而蒸馏塔20c可为经由上部传输线21排出粗丙烯酸以及经由底部传输线23排出高沸点物质(废油)的高沸点分离塔。例如,假设使用7%的丙烷产生60%的转化率,10%的一氧化碳(CO)选择性、10%的二氧化碳(CO2)选择性以及80%的丙烯酸选择性,示出在稳定状态下在POA工艺的各个位置处进料的进料速率。在启动阶段,CO的流速为零,随着反应的进行与清除的CO的量相比产生的CO的量增加更多,由此一氧化碳(CO)的浓度累积。启动阶段进料的组成由7%的丙烷、14%的氧气(O2)、0%的一氧化碳(CO)、59%的二氧化碳(CO2)和20%的水(H2O)组成,而在稳定阶段,进料组成包括7%的丙烷、14%的O2、29.5%的CO、29.5%的CO2和20%的H2O。CO的浓度升高到29.5%。因为CO在工艺期间累积,实际的POA工艺优选地进一步包括通过选择性CO氧化除去CO。如图3中示出可以包括选择性CO氧化的形式提供的实际的POA工艺。可将一氧化碳(CO)氧化单元设置在POA反应器的上游或POA反应器的下游。可将选择性CO去除单元设置在POA反应器的上游,并且可按照以下计算CO去除效果(CO+1/2O2→CO2)。假设CO转化率为87.7%并且使用7%丙烷产生60%的转化率,10%的CO选择性,10%的CO2选择性,80%的丙烯酸选择性,可以计算在稳定状态下包含CO去除的POA工艺中各个位置处进料的进料速率。结果示于以下表3中。在将选择性CO去除单元设置在POA反应器下游的情形中,进料速率稍有不同。本发明的方式通过以下列出并说明的实施例可以获得本发明的更好理解,但是其不理解为限制本发明的范围。本发明的范围由权利要求限定,而且还包含在与权利要求等价的含义以及范围内的所有修改。除非另外提及,以重量基础给出在以下实施例以及对比实施例中指代量的“%”和“份”。制备实施例使用以下水热合成工艺制备基于MoVaTebNbcOx的催化剂。溶液A:将40.11g的仲钼酸铵溶解在400ml热水中,接着溶解7.97g的钒酸铵。然后溶解碲二氢化物(12.0g)。将得到的溶液冷却至35℃。溶液B:将20.62g的草酸二酐以及4.65g的铌酸添加到110ml的水中并加热1小时,由此得到均匀的溶液。将该溶液冷却至35℃。将溶液B加到溶液A中。使用200℃的入口温度以及110℃的出口温度对所述浆液喷雾干燥,由此得到固体产物。在200℃下在空气中煅烧所述固体产物2小时,并在将氮气料流中氧气的量保持在5ppm以下的同时在600℃下烧制2小时。然后研磨所述固体、压碎并筛分。考察60至80目的组分,由其得到Mo1.0V0.3Nb0.12Te0.23Ox催化剂。在此,x为连接到其他元素的氧的量,并取决于其他元素的氧化态。实施例1将丙烷放入具有3/4英寸内径以及包含在以上制备实施例中制备的10g催化剂的不锈钢反应器中并在熔盐中加热。在此,使在以下表1中示出的量的丙烷、氧气、和水蒸气在1400hr-1的空速、360℃以及15psi的条件下经历部分氧化,由此产生含丙烯酸的气体。该含丙烯酸的气体通过从其顶部喷水的吸收塔,被分离为含丙烯酸的溶液和气体副产物。在吸收塔中分离的部分气体副产物被分开并以相当于产生的二氧化碳的量对应的量被除去。而且,与新鲜进料的摩尔比相比,以与由于反应减少的量对应的量进一步添加丙烷和氧气,并由此与气体副产物合并,并将得到的料流供应到固定床反应器中以达到稳定状态。经由传输线使用蒸馏塔将含丙烯酸的溶液分离为丙烯酸溶液和液体副产物。采用气相色谱法分析分离的液体副产物。使分离的丙烯酸溶液经历蒸馏和重结晶,由此产生丙烯酸。在2小时反应时间之后的稳定状态中,回收的气体副产物包含丙烯、未反应的丙烷、一氧化碳、二氧化碳和水蒸气,并在以下表1中示出回收的气体副产物的进料速率以及额外供应的丙烷和氧气。[表1]实施例2该实施例通过图3中示出的工艺来进行。将丙烷放入具有3/4英寸内径以及包含在以上制备实施例中制备的10g催化剂的不锈钢反应器中并在熔盐中加热。在此,使在以下表2中示出的量的丙烷、氧气、和水蒸气在1400hr-1的空速、360℃以及15psi的条件下经历部分氧化,由此产生含丙烯酸的气体。该含丙烯酸的气体通过含Pt催化剂层的氧化单元12,由此一氧化碳被氧化成二氧化碳。而且,含丙烯酸的气体通过从其顶部喷水的吸收塔14,从而被分离为含丙烯酸的溶液和气体副产物。在吸收塔中分离的部分气体副产物被分开并以与产生的二氧化碳的量对应的量被除去。而且,额外供应丙烷和氧气以使其适合新鲜进料的摩尔%,并由此与气体副产物合并,将得到的料流供应到固定床反应器中以达到稳定状态。经由传输线19使用蒸馏塔20将含丙烯酸的溶液分离为丙烯酸溶液和液体副产物。采用气相色谱法分析分离的液体副产物。使分离的丙烯酸溶液经历蒸馏和重结晶,由此产生丙烯酸。在2小时反应时间之后的稳定状态中,回收的气体副产物包含丙烯、未反应的丙烷、一氧化碳、二氧化碳和水蒸气,并在以下表2中示出回收的气体副产物的进料速率以及额外供应的丙烷和氧气。[表2]表2中显而易见的是,在稳定状态中供应到反应器中的一氧化碳(CO)的浓度保持较低,由此不会发生像实施例1的情形中CO浓度累积的问题。因此可以进行安全操作。对比实施例1将丙烷放入具有3/4英寸内径以及包含在以上制备实施例中制备的10g催化剂的不锈钢反应器中并在熔盐中加热。在此,使在以下表3中示出的量的丙烷、氧气、氮气和水蒸气在1400hr-1的空速、360℃以及15psi的条件下经历部分氧化,由此产生含丙烯酸的气体。该含丙烯酸的气体通过从其顶部喷水的吸收塔,从而被分离为含丙烯酸的溶液和气体副产物。在焚化炉中焚烧分离的气体副产物,而含丙烯酸的溶液经由蒸馏塔被分离成丙烯酸溶液和液体副产物。使用气相色谱法计算分离的气体副产物的各个组分的进料速率以及液体副产物。在以下表3中示出结果。总反应时间为2小时。[表3]试验实施例1使用以下等式计算实施例1和2以及对比实施例1的结果。结果示于以下表4中。将丙烷转化率以及丙烯酸选择性以及产率定义如下:–丙烷转化率(%)=(反应的丙烷摩尔数/供应的丙烷摩尔数)×100–丙烯酸选择性(%)=(产生的丙烯酸摩尔数/反应的丙烷摩尔数)×100–丙烯酸产率(%)=(产生的丙烯酸摩尔数/供应的丙烷摩尔数)×100[表4]组分实施例1实施例2对比实施例1转化率%93.597.458.6选择性%80.080.578.0产率%74.878.445.7试验实施例2在相同的条件下进行实施例2和对比实施例1,并将反应时间延长至50小时。作为其结果,在实施例2的条件下,丙烯酸转化率以及选择性没有改变。然而,在对比实施例1中,使用氮气(N2)作为载气,由于在催化剂上的焦炭沉积导致转化率由35%降低至23%。而在实施例2中,仅额外供应在回收的气体副产物中缺乏的氧气和丙烷,从而连续制备丙烯酸。然而,在对比实施例1中,不仅需要连续供应缺乏的氧气和丙烷而且需要连续供应氮气给料。此外,需要额外的步骤分离在反应器中积聚的一氧化碳(CO)和二氧化碳(CO2)。因此,在实施例2中的能量和成本大大低于对比实施例1中的能量和成本。因此,与常规的方法相比,根据本发明的连续制备丙烯酸的方法能够简单地完成并且显著地降低成本。[附图中的附图标记的说明]10:进料传输线,18:回收传输线13、15、17、19、21、22、23:传输线11:部分氧化反应器,12:氧化单元,14:吸收塔,16:控制单元,20:蒸馏塔。当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1