作为突变型异柠檬酸脱氢酶1抑制剂的异噁唑衍生物的制作方法
本发明涉及对于突变型异柠檬酸脱氢酶1(以下也称为“IDH1”)具有优异抑制活性的化合物及其药学上可接受的盐。
背景技术:
异柠檬酸脱氢酶(IDHs:isocitrate dehydrogenases)为将异柠檬酸转化为2-氧代戊二酸(α-酮戊二酸)的一组酶。该组酶可进一步分细为NAD+依赖性异柠檬酸脱氢酶(EC 1.1.1.41)和NADP+依赖性异柠檬酸脱氢酶(EC 1.1.1.42)。
IDH1(异柠檬酸脱氢酶1(NADP+),可溶的)蛋白、IDH2(异柠檬酸脱氢酶2(NADP+),线粒体的)蛋白为分类为NADP+依赖性异柠檬酸脱氢酶(EC 1.1.1.42)的酶。于各种癌中均发现了IDH1基因突变或IDH2基因突变。具体示例包括神经胶质瘤及神经胶质母细胞瘤、急性髓细胞白血病、骨髓增生异常综合征、骨髓增殖性肿瘤、末梢性T细胞性依赖性、软骨肉瘤、骨肉瘤、胆管癌、原始神经外胚层肿瘤、B淋巴母细胞性淋巴瘤、恶性黑色素瘤、前列腺癌、结直肠癌和甲状腺癌等(非专利文献1~16)。
另外,已有报道指出,大部分常常具有多发性软骨肉瘤的奥利埃氏病或马二氏综合征的患者天然保留了IDH1基因突变或IDH2基因突变(非专利文献8,9)。
各种文献报道的共同特征为,IDH1及IDH2的基因突变为点突变,并且突变位点集中于对酶反应而言重要的氨基酸或其附近的氨基酸上。具体而言,IDH1蛋白的132位(以下以R132表示)的精氨酸被另一种氨基酸所取代的突变占IDH1基因突变的大多数。例如,经常发现132位的精氨酸转化为组氨酸(以R132H表示)或转化为胞嘧啶(R132C)、亮氨酸(R132L)、丝氨酸(R132S)、甘氨酸(R132G)、缬氨酸(R132V)等。此外,已知有于G97、R100、H133、A134等产生突变的情况。IDH2基因突变为R140或R172转化为其他氨基酸的突变为大部分。例如已知有R140Q突变或R172K或R172S突变。在大部分的突变情况中还表明,等位基因之一以野生型存在。突变的这些特征表明,突变的IDH1基因及突变的IDH2基因起活化突变的功能。
由IDH1R132H蛋白的功能分析,得知IDH1R132H蛋白具有与野性型的IDH1完全不同的酶活性,即具有将2-氧代戊二酸与NADPH转化为D-2-羟基戊二酸(D-2-hydroxyglutarate:以下称为2-HG)与NADP+的活性(非专利文献17)。在神经胶质瘤或急性髓细胞白血病细胞和具有突变如IDH1的R132H、R132S、R132C、R132G、G97D或IDH2的R140Q或R172K的突变的培养细胞中,可以高浓度产生2-HG。对于其他IDH1突变也有类似的报道(非专利文献18-20)。由这些报道得知,存在如下可能性:肿瘤中表达的突变型IDH1蛋白通过不同于野生型IDH的酶活性所产生的2-HG,可能对肿瘤性质产生影响。
已经表明,诱导表达IDH1R132H的急性髓细胞白血病细胞系TF-1细胞,即使在没有GM-CSF(粒细胞巨噬细胞集落刺激因子(Granulocyte Macrophage Colony-Stimulating Factor))的培养基中也可以生长。还表明,表达IDH1R132H的TF-1细胞通过促红细胞生成素转化红血球的分化受到抑制(非专利文献21)。由这些报道可知,通过突变型IDH1蛋白的功能,可以诱导肿瘤具有如促进的生长或分化的抑制的性质。
同样,在上述奥利埃氏病或马二氏综合征的患者中,也检测到存在高浓度的2-HG。还有报道表明,在部分D-2-羟基戊二酸尿症患者中也检测到IDH1基因突变(非专利文献22)。因此,突变IDH蛋白所产生的2-HG似乎有助于这些疾病的病理。
基于上述,预期抑制突变型IDH1蛋白活性的药物可以用作特异性作用于与IDH1突变有关的疾病例如癌症的治疗药物。
已有报道,酰胺衍生物(专利文献6、7和8)、二环化合物(专利文献9)、氨基吡啶衍生物(专利文献10、11)、氨基嘧啶衍生物(专利文献12)等可以作为抑制突变型IDH1蛋白的活性的化合物。
然而,仍然需要开发对突变型IDH1蛋白具有优异抑制活性的新结构的化合物。
现有技术文献
专利文献
专利文献1:US3336307
专利文献2:WO1994/010145
专利文献3:WO1994/024095
专利文献4:GB2284600
专利文献5:WO2004/072051
专利文献6:WO2012/009678
专利文献7:WO2013/107291
专利文献8:WO2013/107405
专利文献9:WO2012/173682
专利文献10:WO2012/171506
专利文献11:WO2012/171337
专利文献12:WO2013/046136
非专利文献
非专利文献1:H.Yang等人,Clin Cancer Res 18(2012)5562-5571.
非专利文献2:D.W.Parsons等人,Science 321(2008)1807-1812.
非专利文献3:H.Yan等人,N Engl J Med 360(2009)765-773.
非专利文献4:E.R.Mardis等人,N Engl J Med 361(2009)1058-1066.
非专利文献5:P.Paschka等人,J Clin Oncol 28(2010)3636-3643.
非专利文献6:O.Kosmider等人,Leukemia 24(2010)1094-1096.
非专利文献7:A.Tefferi等人,Leukemia 24(2010)1302-1309.
非专利文献8:T.C.Pansuriya等人,Nat Genet 43(2011)1256-1261.
非专利文献9:M.F.Amary等人,Nat Genet 43(2011)1262-1265.
非专利文献10:D.R.Borger等人,Oncologist 17(2012)72-79.
非专利文献11:T.Shibata等人,Am J Pathol 178(2011)1395-1402.
非专利文献12:M.R.Kang等人,Int J Cancer 125(2009)353-355.
非专利文献13:T.Sjoblom等人,Science 314(2006)268-274.
非专利文献14:J.P.Hemerly等人,Eur J Endocrinol 163(2010)747-755.
非专利文献15:R.A.Cairns等人,Blood 119(2012)1901-1903.
非专利文献16:X.Liu等人,Cancer Med 2(2013)803-814.
非专利文献17:L.Dang等人,Nature 462(2009)739-744.
非专利文献18:P.S.Ward等人,Cancer Cell 17(2010)225-234.
非专利文献19:S.Gross等人,J Exp Med 207(2010)339-344.
非专利文献20:P.S.Ward等人,Oncogene 31(2012)2491-2498.
非专利文献21:J.A.Losman等人,Science 339(2013)1621-1625.
非专利文献22:L.E.Vissers等人,Am J Med Genet A 155A(2011)2609-2616.
技术实现要素:
技术问题
本发明系有鉴于上述作出,本发明的目的为提供对突变型IDH1蛋白具有优异抑制活性的新结构的化合物。
技术手段
为实现上述目的,本发明人合成了具有各种结构的化合物,并测试了它们对突变型IDH1蛋白的抑制活性。结果发现,具有异唑骨架的特定化合物对于突变型IDH1蛋白具有优异抑制活性,通过该蛋白可抑制2-HG的产生,同时该化合物还可以有效地抑制表达该蛋白的各种肿瘤的生长,基于这些发现,完成了本发明。
因此,本发明涉及具有异唑骨架并且对于突变型IDH1蛋白具有抑制活性的化合物、其药学上可接受的盐及其用途,更具体而言,本发明提供:
[1]通式(I)的化合或其药学上可接受的盐:
式1
式中,
Z-Y表示N-O或O-N,
R1表示任选具有1至3个独立选自下述A组的取代基的苯基,或表示任选具有1至3个独立选自下述A组的取代基的吡啶基;
R2表示-NR21R22、任选具有1至3个独立选自下述B组的取代基的C1~C6烷基、任选具有1至3个独立选自下述C组的取代基的C3~C6环烷基,或者于环内具有1或2个独立选自氮原子和氧原子的杂原子的4员至6员杂环基,
该4员至6员杂环基任选具有1至3个独立选自下述C组的取代基,且
于该杂环内任选连接桥接结构,或于该杂环上任选螺结合1个C3~C6环烷基环,
R21及R22分别独立表示氢原子、C1~C6烷基或-C(=O)R23,
R23表示C2~C6烯基或C2~C6炔基,
R3表示下述式(II)至式(IV)之一:
式2
式中,
R31表示氢原子、卤素原子、1至3个卤素原子所取代的C1~C6烷基、C3~C6环烷基或C1~C6烷基羰基,
R32表示氢原子或C1~C6烷基,或
R31及R32任选一起形成环己烷环,
R33表示氢原子或C1~C6烷基,或
R32及R33任选一起形成环丙烷环、
R34表示氢原子或C1~C6烷基,
R35表示C1~C6烷基,
R36表示氢原子或C1~C6烷基,
R37表示氢原子或C1~C6烷基,或
R36及R37任选一起形成苯环、
R38表示氢原子或卤素原子,
X表示氮原子或CH,
W表示氮原子或CH,
虚线表示单键或双键。
所述化合物或其药学上可接受的盐。
A组:卤素原子、C1~C6烷基和C1~C6烷氧基
B组:卤素原子、羟基、C1~C6烷氧基、C1~C6烷基氨基和二C1~C6烷基氨基
C组:C2~C6烯基、卤素原子、羟基、氰基、任选具有1至3个独立选自下述D组的取代基的C1~C6烷基、C1~C6烷氧基、-NR211R212、-C(=O)R213和-SO2R213
R211及R212分别独立表示氢原子或C1~C6烷基,
R213表示C2~C6烯基或C2~C6炔基。
D组:氨基、C1~C6烷氧基、二C1~C6烷基氨基、氧代基和C3~C6环烷基
[2][1]所述的化合物或其药学上可接受的盐,在式(I)中,R1表示任选具有1至3个独立地选自上述A组的取代基的苯基。
[3][1]或者[2]所述的化合物或其药学上可接受的盐,在式(I)中,
R2表示任选具有1至3个独立选自上述B组的取代基的C1~C6烷基,或环内具有1或2个独立选自氮原子和氧原子的杂原子的4至6员的脂肪族杂环基,该4至6员的脂肪族杂环基任选具有1至3个独立选自上述C组的取代基。
[4][1]至[3]中任一项所述的化合物或者其药学上可接受的盐,在式(I)中,R2表示下列之一:
式3
[5][1]至[4]中任一项所述的化合物或其药学上可接受的盐,在式(I)中,
R3表示下述式(IV)或式(V):
式4
在式(IV)及式(V)中,
R3a表示氢原子或任选由1至3个卤素原子所取代的C1~C6烷基,
R3b表示氢原子或C1~C6烷基,
R3c表示氢原子或C1~C6烷基,
R3d表示氢原子或C1~C6烷基,
R3e表示氢原子或卤素原子,且
虚线表示单键或双键。
[6][1]至[5]中任一项所述的化合物或者其药学上可接受的盐,在式(I)中,R3表示下列之一
式5
[7]通式VI化合物或其药学上可接受的盐:
式6
式中,
R4、R5及R6分别独立表示氢原子或卤素原子,
R7表示下列之一,
式7
R8及R9分别独立表示氢原子或C1~C6烷基,
[8]通式VII化合物或其药学上可接受的盐:
式8
式中,
R10、R11及R12分别独立表示氢原子或卤素原子,
R13表示下列之一,
式9
R14分别独立表示氢原子或C1~C6烷基。
[9]选自下述任一个的化合物或其药学上可接受的盐。
(2E)-3-(1-{[5-(3-甲基氧杂环丁烷-3-基)-3-(2,4,6-三氯苯基)-1,2-唑-4-基]羰基}-1H-吲哚-4-基)丙-2-烯酸
(2E)-3-(1-{[5-(2-氟丙烷-2-基)-3-(2,4,6-三氯苯基)-1,2-唑-4-基]羰基}-3-甲基-1H-吲哚-4-基)丙-2-烯酸
(2E)-3-(1-{[5-(叔丁基)-3-(2,4,6-三氯苯基)-1,2-唑-4-基]羰基}-1H-吲哚-4-基)丙-2-烯酸
(2E)-3-(1-{[3-(2,4-二氯-6-氟苯基)-5-(2-氟丙烷-2-基)-1,2-唑-4-基]羰基}-3-甲基-1H-吲哚-4-基)丙-2-烯酸
(2E)-3-(1-{[3-(2,4-二氯苯基)-5-(2-氟丙烷-2-基)-1,2-唑-4-基]羰基}-3-甲基-1H-吲哚-4-基)丙-2-烯酸
(2E)-[1-{[5-(1-丙烯酰基哌啶-4-基)-3-(2,4,6-三氯苯基)-1,2-唑-4-基]羰基}-3,4-二氢苯并[cd]吲哚-5(1H)-亚基]乙酸
(2E)-3-(1-{[5-(1-丙烯酰基哌啶-4-基)-3-(2,4,6-三氯苯基)-1,2-唑-4-基]羰基}-3-甲基-1H-吲哚-4-基)丙-2-烯酸
(2E)-[1-{[5-(1-丙烯酰基哌啶-4-基)-3-(2,4-二氯-6-氟苯基)-1,2-唑-4-基]羰基}-3,4-二氢苯并[cd]吲哚-5(1H)-亚基]乙酸
(2E)-[1-{[5-(1-丙烯酰基哌啶-4-基)-3-(2,4-二氯-5-氟苯基)-1,2-唑-4-基]羰基}-3,4-二氢苯并[cd]吲哚-5(1H)-亚基]乙酸
(2E)-[1-{[5-(1-丙烯酰基哌啶-4-基)-3-(2,4-二氯苯基)-1,2-唑-4-基]羰基}-3,4-二氢苯并[cd]吲哚-5(1H)-亚基]乙酸
[10](2E)-3-(1-{[5-(2-氟丙烷-2-基)-3-(2,4,6-三氯苯基)-1,2-唑-4-基]羰基}-3-甲基-1H-吲哚-4-基)丙-2-烯酸或者其药学上可接受的盐。
[11](2E)-3-(1-{[5-(2-氟丙烷-2-基)-3-(2,4,6-三氯苯基)-1,2-唑-4-基]羰基}-3-甲基-1H-吲哚-4-基)丙-2-烯酸叔丁基胺盐。
[12](2E)-[1-{[5-(1-丙烯酰基哌啶-4-基)-3-(2,4,6-三氯苯基)-1,2-唑-4-基]羰基}-3,4-二氢苯并[cd]吲哚-5(1H)-亚基]乙酸或者其药学上可接受的盐。
[13](2E)-[1-{[5-(1-丙烯酰基哌啶-4-基)-3-(2,4,6-三氯苯基)-1,2-唑-4-基]羰基}-3,4-二氢苯并[cd]吲哚-5(1H)-亚基]乙酸叔丁基胺盐。
[14]突变型异柠檬酸脱氢酶1抑制剂,包括将[1]至[13]中任一项所述的化合物或者其药学上可接受的盐作为活性成分。
[15]D-2-羟基戊二酸产生抑制剂,包括将[1]至[13]中任一项所述的化合物或者其药学上可接受的盐作为活性成分。
[16]药物组合物,包括将[1]至[13]中任一项所述的化合物或者其药学上可接受的盐作为活性成分。
[17]对抗具有突变型异柠檬酸脱氢酶1基因突变的肿瘤的抗肿瘤剂,包括将[1]至[13]中任一项所述的化合物或者其药学上可接受的盐作为活性成分。
[18][17]所述的抗肿瘤剂,其中所述肿瘤为脑瘤(包括神经胶质瘤)、急性髓细胞白血病、骨髓增生异常综合征、骨髓增殖性肿瘤、外周T细胞淋巴瘤、软骨肉瘤、骨肉瘤、胆管癌、原始神经外胚层肿瘤、B淋巴母细胞性淋巴瘤、恶性黑色素瘤、前列腺癌、结直肠癌或甲状腺癌。
[19][1]至[13]中任一项所述的化合物或者其药学上可接受的盐,用于治疗具有突变型异柠檬酸脱氢酶1基因突变的肿瘤的治疗方法中。
[20][19]所述的化合物或者其药学上可接受的盐,其中所述肿瘤为脑瘤(包括神经胶质瘤)、急性髓细胞白血病、骨髓增生异常综合征、骨髓增殖性肿瘤、外周T细胞淋巴瘤、软骨肉瘤、骨肉瘤、胆管癌、原始神经外胚层肿瘤、B淋巴母细胞性淋巴瘤、恶性黑色素瘤、前列腺癌、结直肠癌或甲状腺癌。
[21][1]至[13]中任一项所述的化合物或者其药学上可接受的盐在生产具有突变型异柠檬酸脱氢酶1基因突变的肿瘤的药物组合物中的用途。
[22][21]所述的用途,其中所述肿瘤为脑瘤(包括神经胶质瘤)、急性髓细胞白血病、骨髓增生异常综合征、骨髓增殖性肿瘤、外周T细胞淋巴瘤、软骨肉瘤、骨肉瘤、胆管癌、原始神经外胚层肿瘤、B淋巴母细胞性淋巴瘤、恶性黑色素瘤、前列腺癌、结直肠癌或甲状腺癌。
[23]治疗具有突变型异柠檬酸脱氢酶1基因突变的肿瘤的方法,该方法包括给予[1]至[13]中任一项所述的化合物或者其药理上可接受的盐。
[24][23]所述的治疗方法,其中所述肿瘤为脑瘤(包括神经胶质瘤)、急性髓细胞白血病、骨髓增生异常综合征、骨髓增殖性肿瘤、外周T细胞淋巴瘤、软骨肉瘤、骨肉瘤、胆管癌、原始神经外胚层肿瘤、B淋巴母细胞性淋巴瘤、恶性黑色素瘤、前列腺癌、结直肠癌或甲状腺癌。
迄今为止,已经发现,具有异唑骨架的化合物可以作为抗炎剂(专利文献1)、多巴胺受体亚型配体(专利文献2)、免疫抑制剂(专利文献3)、除草剂(专利文献4)、热激蛋白抑制剂(专利文献5)的报道,但是尚无关于抑制突变型IDH蛋白活性的报道。
本发明的有利技术效果
本发明的化合物或其药学上可接受的盐对于突变型IDH1蛋白具有强的活性抑制作用,因此可以抑制表达该蛋白的细胞产生2-HG并抑制其生长。已经在各种肿瘤中发现了IDH1蛋白的突变,所以本发明的化合物或其药学上可接受的盐尤其是可以作为抗肿瘤剂。
附图说明
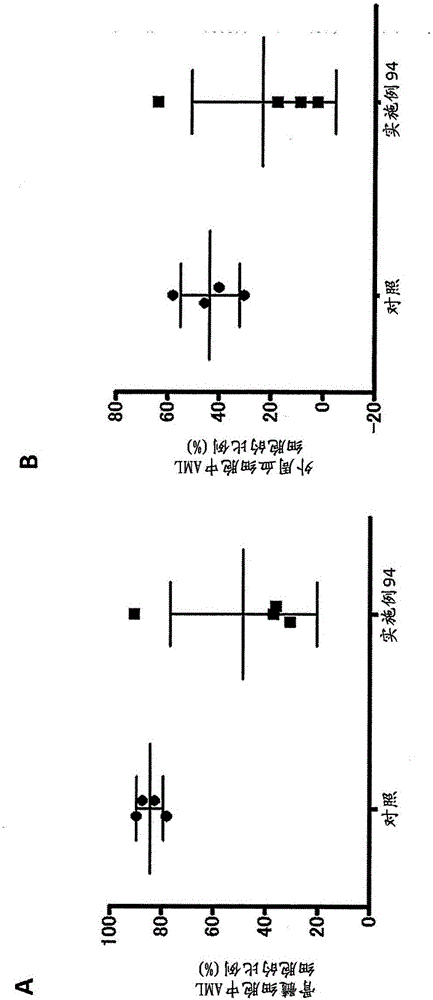
图1表示在荷载包括IDH1R132H的4个基因的急性髓细胞白血病(AML)小鼠模型中,测定本发明化合物的2-HG产生抑制活性的结果示意图。图1A表示血浆中2-HG的浓度,图1B表示骨髓细胞中2-HG的量。
图2表示在荷载包括IDH1R132H的4个基因的急性髓细胞白血病(AML)小鼠模型中,测定本发明化合物的抗肿瘤活性的结果示意图。图2A表示骨髓细胞中的EGFP阳性AML细胞的比例,图2B表示外周血细胞中EGFP阳性AML细胞的比例。也示出了平均值±分散值。
图3表示在植入了IDH1/R132H突变型神经胶质母细胞瘤A1074的小鼠模型中测定本发明化合物的抗肿瘤活性的结果示意图。也示出了本试验中各组肿瘤体积的平均值和标准差。
图4表示在植入了IDH1/R132H突变型神经胶质母细胞瘤A1074的小鼠模型中测定本发明的化合物的抗肿瘤活性的结果示意图。也示出了给予混合饲料4周后肿瘤的重量。
具体的实施方式
本发明提供下述通式(I)所示化合物或其药学上可接受的盐。
式10
式中,Z-Y表示N-O或O-N。
式中,R1表示任选具有1至3个独立选自下述A组的取代基的苯基,或任选具有1至3个独立选自下述A组的取代基的吡啶基。优选R1表示任选具有1至3个独立选自下述A组的取代基的苯基。
A组:卤素原子、C1~C6烷基和C1~C6烷氧基。
本发明中,“卤素原子”为氟原子、氯原子、溴原子或碘原子。
本发明中,“C1~C6烷基”为具有1至6个碳原子的直链或支链烷基。示例包括甲基、乙基、丙基、异丙基、丁基、异丁基、s-丁基、t-丁基、戊基、异戊基、2-甲基丁基、新戊基、1-乙基丙基、己基、异己基或4-甲基戊基。
本发明中,“C1~C6烷氧基”表示氧基团与上述C1-C6烷基连接的基团,示例包括连接甲氧基、乙氧基、n-丙氧基、异丙氧基、丁氧基、异丁氧基、s‐丁氧基、t‐丁氧基、戊氧基、异戊氧基、2‐甲基丁氧基、己氧基或异己基氧基。
式中,R2表示-NR21R22、任选具有1至3个独立选自下述B组的取代基的C1~C6烷基、任选具有1至3个独立选自下述C组的取代基的C3~C6环烷基,或环内具有1或者2个独立选自氮原子及氧原子的杂原子的4员至6员杂环基。
在本发明中,“-NR21R22”中的R21及R22分别独立表示氢原子、C1~C6烷基或-C(=O)R23。R23表示C2~C6烯基或C2~C6炔基。“4员至6员杂环基”任选具有1至3个独立选自下述C组的取代基,且该杂环中任选连接桥接结构,或于该杂环上具有1个通过螺连接合的C3~C6环烷基环。
B组:卤素原子、羟基、C1~C6烷氧基、C1~C6烷基氨基和二C1~C6烷基氨基
C组:C2~C6烯基、卤素原子、羟基、氰基、任选具有1至3个独立选自下述D组的取代基的C1~C6烷基、C1~C6烷氧基、-NR211R212、-C(=O)R213和-SO2R213
在本发明中,“-NR211R212”中的R211及R212分别独立表示氢原子或C1~C6烷基。“-C(=O)R213”或“-SO2R213”中的R213表示C2~C6烯基或C2~C6炔基。
D组:氨氨基、C1~C6烷氧基、二C1~C6烷基氨基、氧代基和C3~C6环烷基
在本发明中,“C3~C6环烷基”为环丙基、环丁基、环戊基或环己基。
在本发明中,“C1~C6烷基氨基”为被1个上述C1~C6烷基取代的氨氨基。示例包括甲基氨氨基、乙基氨氨基、丙基氨氨基、异丙基氨氨基、丁基氨氨基、异丁基氨氨基、s‐丁基氨氨基、t‐丁基氨氨基、戊基氨氨基、异戊基氨氨基、2‐甲基丁基氨氨基、新戊基氨氨基、1‐乙基丙基氨氨基、己基氨氨基或异己基氨氨基等。
在本发明中,“二C1~C6烷基氨基”表示被2个相同或相异的上述C1~C6烷基所取代的氨氨基。示例包括二甲基氨基、二乙基氨基、二丙基氨基、二异丙基氨基、二丁基氨基、二异丁基氨基、二戊基氨基、二新戊基氨基、二己基氨基、N‐乙基-N-甲基氨基、N‐甲基-N-丙基氨基、N-异丙基-N-甲基氨基、N-丁基-N-甲基氨基、N-异丁基-N-甲基氨基、N-乙基-N-丙基氨基、N-乙基-N-异丙基氨基、N-丁基-N-乙基氨基或N-乙基-N-异戊基氨基。
在本发明中,“C2~C6烯基”表示在分子中具有1个双键和2至6个碳原子的直链或支链烯基,示例包括乙烯基、烯丙基或异丙烯基。
在本发明中,“C2~C6炔基”为在分子中具有1个叁键和2至6个碳原子的直链或支链C2-C6炔基,示例包括乙炔基、丙-1-炔基、丙-2-炔基或丁-3-炔基。
在本发明中,“杂环基”表示除碳外,在环中还含有1或2个独立选自氮原子及氧原子的原子的单环芳族或脂肪族化合物。示例包括呋喃基、吡咯基、唑基、异唑基、咪唑基、咪唑基、吡啶基、吡嗪基、嘧啶基、哒嗪基、环氧乙烷基、氮丙啶基、氧杂环丁烷基、氮杂环丁基、四氢呋喃基、吡咯烷基、四氢吡喃基、哌嗪基、四氢噻喃基、吗啉子基、吗啉基或哌嗪基。
优选R2为任选具有1至3个独立选自B组的取代基的C1~C6烷基,或环内具有1或者2个选自氮原子及氧原子的杂原子的4员至6员的杂环基。
更优选R2为下式之一。
式11
式中,R3表示下述式(II)至式(IV)之一。
式12
式中,R31表示氢原子、卤素原子、任选被1至3个卤素原子所取代的C1~C6烷基、C3~C6环烷基或C1~C6烷基羰基,
R32表示氢原子或C1~C6烷基,或
R31及R32一起共同形成环己烷环,
R33表示氢原子或C1~C6烷基,或
R32及R33为R32一起共同形成环丙烷环,
R34表示氢原子或C1~C6烷基,
R35表示C1~C6烷基,
R36表示氢原子或C1~C6烷基,
R37表示氢原子或C1~C6烷基,或
R36及R37一起共同形成苯环、
R38表示氢原子或卤素原子,
X表示氮原子或CH,
W表示氮原子或CH,
虚线表示单键或双键。
在本发明中,“C1~C6烷基羰基”表示羰基与上述C1-C6烷基连接的基团。示例包括连接甲基羰基、乙基羰基、n-丙基羰基或异丙基羰基。
优选R3表示下述式(IV)或下述式(V)。
式13
在式(IV)及式(V)中,
R3a表示氢原子或任选由1至3个卤素原子所取代的C1~C6烷基,
R3b表示氢原子或C1~C6烷基,
R3c表示氢原子或C1~C6烷基,
R3d表示氢原子或C1~C6烷基,
R3e表示氢原子或卤素原子,
虚线表示单键或双键。
更优选R3表示下列之一。
式14
更优选通式I所代表的化合物为下述通式(VI)或下述通式(VII)所代表的化合物。
式15
式中,
R4、R5及R6分别独立表示氢原子或卤素原子,
R7表示下列之一,
式16
R8及R9分别独立表示氢原子或C1~C6烷基
式17
式中,
R10、R11及R12分别独立表示氢原子或卤素原子,
R13表示下列之一,
式18
R14分别独立表示氢原子或C1~C6烷基
特别优选通式(I)所表示的化合物选自下述的任一种化合物。
(2E)-3-(1-{[5-(3-甲基氧杂环丁烷-3-基)-3-(2,4,6-三氯苯基)-1,2-唑-4-基]羰基}-1H-吲哚-4-基)丙-2-烯酸
(2E)-3-(1-{[5-(2-氟丙烷-2-基)-3-(2,4,6-三氯苯基)-1,2-唑-4-基]羰基}-3-甲基-1H-吲哚-4-基)丙-2-烯酸
(2E)-3-(1-{[5-(叔丁基)-3-(2,4,6-三氯苯基)-1,2-唑-4-基]羰基}-1H-吲哚-4-基)丙-2-烯酸
(2E)-3-(1-{[3-(2,4-二氯-6-氟苯基)-5-(2-氟丙烷-2-基)-1,2-唑-4-基]羰基}-3-甲基-1H-吲哚-4-基)丙-2-烯酸
(2E)-3-(1-{[3-(2,4-二氯苯基)-5-(2-氟丙烷-2-基)-1,2-唑-4-基]羰基}-3-甲基-1H-吲哚-4-基)丙-2-烯酸
(2E)-[1-{[5-(1-丙烯酰基哌啶-4-基)-3-(2,4,6-三氯苯基)-1,2-唑-4-基]羰基}-3,4-二氢苯并[cd]吲哚-5(1H)-亚基]乙酸
(2E)-3-(1-{[5-(1-丙烯酰基哌啶-4-基)-3-(2,4,6-三氯苯基)-1,2-唑-4-基]羰基}-3-甲基-1H-吲哚-4-基)丙-2-烯酸
(2E)-[1-{[5-(1-丙烯酰基哌啶-4-基)-3-(2,4-二氯-6-氟苯基)-1,2-唑-4-基]羰基}-3,4-二氢苯并[cd]吲哚-5(1H)-亚基]乙酸
(2E)-[1-{[5-(1-丙烯酰基哌啶-4-基)-3-(2,4-二氯-5-氟苯基)-1,2-唑-4-基]羰基}-3,4-二氢苯并[cd]吲哚-5(1H)-亚基]乙酸
(2E)-[1-{[5-(1-丙烯酰基哌啶-4-基)-3-(2,4-二氯苯基)-1,2-唑-4-基]羰基}-3,4-二氢苯并[cd]吲哚-5(1H)-亚基]乙酸
最优选本发明的化合物为(2E)-3-(1-{[5-(2-氟丙烷-2-基)-3-(2,4,6-三氯苯基)-1,2-唑-4-基]羰基}-3-甲基-1H-吲哚-4-基)丙-2-烯酸或者其药学上可接受的盐,或者(2E)-[1-{[5-(1-丙烯酰基哌啶-4-基)-3-(2,4,6-三氯苯基)-1,2-唑-4-基]羰基}-3,4-二氢苯并[cd]吲哚-5(1H)-亚基]乙酸或者其药学上可接受的盐。优选这些化合物的药学上可接受的盐为叔丁基胺盐(t-丁基胺盐)。
在本发明中,“药学上可接受的盐”指不具有显著毒性并且可在药物组合物中使用的盐。本当具有碱性基团时,发明的化合物可通过与酸反应形成盐,当具有酸性基时,可通过与碱反应而成为盐。基于碱性基团的盐,示例包括但不限于氢卤,例如氢氟酸盐、盐酸盐、氢溴酸盐、氢碘酸盐;无机酸盐,如硝酸盐、过氯酸盐、硫酸盐和磷酸盐;烷基磺酸盐,如甲磺酸盐、三氟甲磺酸盐、乙磺酸盐;芳基磺酸盐,如苯磺酸盐、p-甲苯磺酸盐;和羧酸盐,如乙酸盐、草酸盐、酒石酸盐和马来酸盐。
另一方面,基于酸性基团的盐的示例包括但不限于:金属盐,包括碱金属盐,如钠盐、钾盐和锂盐;碱土金属盐,如钙盐和镁盐以及其他盐,如铝盐和铁盐;胺盐,包括无机盐,如t-丁基胺盐、t-辛基胺盐、二苯甲基胺盐、吗啉盐、葡萄糖胺盐、苯基甘氨酸烷基酯盐、乙二胺盐、N-甲基葡糖胺盐、胍盐、二乙基胺盐、三乙基胺盐、二环己基胺盐、N,N’-二苯甲基乙二胺盐、氯普鲁卡因盐、普鲁卡因盐、二乙醇胺盐、N-苯甲基苯乙基胺盐、哌嗪盐、四甲基铵盐和三(羟基甲基)氨氨基甲烷盐;及氨基酸盐,如甘氨酸盐、赖氨酸盐、精氨酸盐、鸟氨酸盐、谷氨酸盐和天冬氨酸盐。
本发明的化合物或者其药学上可接受的盐当放置在大气中或者再结晶时,可能掺入水分子成为水合物,这种水合物也包含于本发明的盐中。
本发明的化合物或者其药学上可接受的盐当置于溶剂中或者再结晶时,可能掺入某种溶剂而成为溶剂合物,这种溶剂合物也包含于本发明的盐中。
本发明的化合物或者其药学上可接受的盐包括所有的异构体(非对映异构体、光学异构体、几何异构体、旋转异构体)。
对于本发明的化合物,这些异构物及这些异构物的混合物皆以单一式表示。因此,本发明包括所有这些异构物及这些异构物的任意比例的混合物。
本发明还提供突变型IDH1抑制剂,该突变型IDH1抑制剂包括将上述本发明化合物或者其药学上可接受的盐作为活性成分突变。在本发明中,“突变型IDH1”中的突变的示例包括但不限于IDH1的第132位的精氨酸(以下以R132表示)的突变、G97的突变、R100的突变、H133的突变和A134的突变。R132突变的示例包括但不限于:组氨酸的突变(R132H)、胞嘧啶的突变(R132C)、亮氨酸的突变(R132L)、丝氨酸的突变(R132S)、甘氨酸的突变(R132G)和缬氨酸的突变(R132V)。本发明的化合物或者其药学上可接受的盐特别适合作为IDH1的R132突变体的抑制剂。
衍生自人野生型IDH1的典型氨基酸序列记载于Genebank的NP_005887.2或UniprotKB的O75874。
已经发现,特定的突变型IDH1,具有将2-氧代戊二酸与NADPH转化为2-HG与NADP+的酶活性,这是野生型IDH1所不具有的,并且在具有特定的突变型IDH1的细胞中也突变确认2-HG以高浓度产生(非专利文献17-20)。在本试验实施例(试验实施例1、2、3和5)中,发现本发明的化合物通过抑制该酶活性,能够抑制2-HG的产生。因此,本发明提供2-HG产生抑制剂,包括将上述本发明的化合物或者其药学上可接受的盐作为活性成分。
可以将本发明的化合物或者其药学上可接受的盐制备为药物组合物,也可知制备为研究目的的试剂。
在构成本发明化合物的一个或多个原子上,本发明的化合物也可以含有原子非天然比例的原子同位素。原子同位素的示例包括氘(2H)、氚(3H)、碘-125(125I)或碳-14(14C)。本发明的化合物还可以是同位素标记的,例如用同位素氚(3H)、碘-125(125I)或碳-14(14C)进行标记。标记的同位素可以用作治疗或预防试剂、研究试剂(例如分析试剂)和诊断剂(例如活体内图像诊断剂)使用。本发明的化合物的所有同位体突体无论是否具有放射性,均包含在本发明的范围中。
在患有脑瘤(包括神经胶质瘤)、急性髓细胞白血病、骨髓增生异常综合征、骨髓增殖性肿瘤、外周T细胞淋巴瘤、软骨肉瘤、骨肉瘤、胆管癌、原始神经外胚层肿瘤、B淋巴母细胞性淋巴瘤、恶性黑色素瘤、前列腺癌、结直肠癌和甲状腺癌等癌的患者,或者患有奥利埃氏病或马二氏症候群的患者中,发现了IDH1基因突变或IDH2基因突变(非专利文献1-16)。此外,在本试验实施例(试验实施例4和7)中,发现本发明的化合物可抑制各种肿瘤细胞的增殖。因此,含有作为活性成分的本发明化合物或者其药学上可接受的盐的药物组合物可以例如靶向治疗这些疾病,特别适合作为抗肿瘤剂。
对于IDH1基因突变是否存在可以通过使用本领域中已知的方法在患者的试验组织(例如通过采血或生检等进行收集)中进行确认,所述方法例如免疫印迹法、ELISA、DNA芯片、FISH分析、组织免疫染色、所属领域公知的其他基因分析法(例如,桑格序列(Sanger sequence)分析、次代DNA序列分析(NGS)、PCR、LCR(连接酶链反应)、SDA(链替代扩增(Strand displacement amplification))、NASBA(依赖核酸序列的扩增)、ICAN(恒温和嵌合引物引发的核酸扩增(Isothermal and chimeric primer-initiated amplification))和LAMP法(环介导的等温扩增(Loop-mediated isothernal amplification))等}等或者病理学方法。
在本发明中,所述“肿瘤”不限于恶性肿瘤,可以包括所有种类的肿瘤。其示例包括癌、肉瘤和良性肿瘤等。特别是,恶性肿瘤也表示为“癌”。
本发明的抗肿瘤剂可以与其他抗肿瘤剂或其他治疗方法(例如放疗或免疫疗法)组合使用。
其他抗肿瘤剂的示例包括烷化剂、各种抗代谢物、抗肿瘤抗生素、抗肿瘤植物成分、BRM(生物学应答性调节剂)、激素、维生素、抗肿瘤抗体、分子靶向药物和其他抗肿瘤药物。
更具体地讲,烷化剂的示例包括:烷化剂,如氮芥类、氮芥N-氧化物或者苯丁酸氮芥;氮杂环丙烷类烷化剂,如卡波醌(Carboquone)或者噻替哌;环氧化物类烷化剂,如二溴甘露醇或者二溴卫矛醇;亚硝脲类烷化剂,如卡莫司汀、洛莫司汀、司莫司汀、尼莫司汀盐酸盐、链脲佐菌素、氯脲霉素或者雷莫司汀;其它如二甲磺酸丁酯、甲苯磺酸甲磺丙胺或达卡巴嗪。
各种抗代谢物的示例包括:嘌呤抗代谢物,如6-巯基嘌呤、6-硫代鸟嘌呤或者硫磺菊素;嘧啶抗代谢物,如氟尿嘧啶、替加氟、替加氟·尿嘧啶、卡莫氟、脱氧氟尿苷、溴尿核苷、阿糖胞苷或者依诺他滨;叶酸抗代谢物,如甲氨蝶呤或者三甲氨蝶呤。
抗肿瘤抗生素的示例包括:蒽环类抗生素抗肿瘤药物,如丝裂霉素C、博莱霉素、培普利欧霉素、柔红霉素、阿柔比星、多柔比星、吡柔比星、THP-阿霉素、4’-表多柔比星和表柔比星;其它如色霉素A3或放线菌素D。
抗肿瘤植物成分的示例包括:长春花生物碱类,如长春酰胺、长春新碱或者长春花碱;紫杉烷类,如紫杉醇、多西他赛;和表鬼臼毒素类,如依托泊苷或替尼泊苷。
BRM的示例包括肿瘤坏死因子或吲哚美辛。
激素的示例包括氢化可的松、地塞米松、甲基泼尼松龙、泼尼松龙、普拉睾酮、倍他米松、曲安奈德、羟甲烯龙、诺龙、美替诺龙、磷雌酚、乙炔基雌甾二醇、氯地孕酮或醋酸甲羟孕酮。
维生素的示例包括维他命C或维他命A。
抗肿瘤抗体和分子靶向药物的示例包括赫塞汀、利妥昔单抗、西妥昔单抗、尼妥珠单抗、狄诺塞麦、贝伐单抗、英夫利昔单抗、伊马替尼、吉非替尼、厄洛替尼、舒尼替尼、拉帕替尼和索拉非尼。
其他抗肿瘤药物的示例包括顺铂、卡铂、奥沙利铂、他莫昔芬、喜树碱、异环磷酰胺、环磷酰胺、美法仑、L-门冬酰胺酶、醋葡醛内酯、西索菲兰、溶链菌、甲基芐肼、哌泊溴烷、新制癌菌素、羟基脲、乌苯美司和云芝多糖。
本发明的化合物或者其药学上可接受的盐可以与具有增强治疗有益特性的辅助部分缀合。典型的有用特性的示例包括促进对于靶向区域(例如肿瘤)的化合物的传递、对于靶向区域中的化合物的治疗浓度的维持、改变化合物的药代动力学或药效动力学特性或改善化合物的治疗指数或安全性。例如,抗体特异性识别的靶区域或在靶区域表达的受体的配体可以作为适当的辅助部分。
在将本发明的化合物或者其药学上可接受的盐作为药物组合物进行配制时,使用的药学上可接受的载体的示例包括但不限于无菌水、生理盐水、植物油、溶剂、基质、乳化剂、悬浮剂、表面活性剂、稳定剂、香味剂、芳香剂、赋形剂、载体、防腐剂、粘合剂、稀释剂、等张剂、安慰剂、膨胀剂、崩解剂、缓冲剂、包衣剂、润滑剂、着色剂、甜味剂、黏稠剂、矫味矫臭剂、增溶剂或者其他添加剂。本发明的化合物或者其药学上可接受的盐可以根据治疗目的等配制成各种形式,例如片剂、散剂、颗粒剂、胶囊剂和溶液剂。本发明的化合物或者其药学上可接受的盐也可以例如以脂质体传递系统的形式给药。前述能够增强治疗有益特性的辅助部分(例如抗体或配体)可以添加到该脂质体中。
对患者的给药可以为口服给药或者为胃肠外给药。胃肠外给药的示例包括静脉给药、动脉内给药、肌肉内给药、胸腔内给药、腹腔内给药、靶向部位(例如肿瘤)的直接给药。
只要给药量是用于治疗目的疾病的有效量则给药量无需特别限定。可以根据患者的年龄、体重、症状、健康状态、疾病进展程度适宜地选择给药量。给药频度并无特别限制,可以根据目的做适宜选择。例如,日剂量可以每天1次给药,或者其可以分为两个或多个部分分次给药。将本发明的药物给药于人类时,活性成分的剂量范围通常为每天约0.01mg/kg体重~约500mg/kg体重,优选为每天约0.1mg/kg体重~约100mg/kg体重。对人类进行给药时,优选日剂量为每日给药1次,或者分为2~4次进行给药,优选给药以适当间隔重复进行。
在制备作为药物的本发明的化合物或者其药学上可接受的盐时,该药物可以任选含有该药物可接受的其它成分,例如无菌水或生理盐水、缓冲剂和防腐剂。该药物可以以适合于接受者(例如细胞或其分级物、组织、实验动物)的剂量给药,目的在于适合于例如抑制突变型IDH1、抑制2-HG的产生或者抑制肿瘤的生长。
其次,描述了通式(I)所述化合物的代表性生产方法。本发明的化合物可由各种生产方法所生产。以下所示生产方法仅用于说明的目的。应当理解,本发明并非受限于这些实施例。通式(I)所述化合物以及用于生产它们的中间体可以利用以下所示各种公知反应而生产。在这方面,原料或中间体的各种官能团可以通过适当的保护基团进行保护。此类官能团的示例包括羟基、羧基、氨基。保护基团的种类以及这些保护基团的导入与除去的条件例如可参考“有机合成中的保护基团“(Protective Groups in Organic Synthesis)(T.W.Green and P.G.M.Wuts,John Wiley&Sons,Inc.,New York,2006)。
[生产方法1]
在式(1)所述化合物中,以下所述化合物1a例如可通过下述反应流程生产:
式19
其中,R1、R31、R32、R33、R38、W、X、Y和Z如上文所定义。其中,虚线表示单键或双键。R39表示羧基的保护基。
R24表示任选具有1至3个独立选自下述B组的取代基的C1~C6烷基、任选具有1至3个独立选自下述C组的取代基的C3~C6环烷基或环内具有1个氧原子的4员至6员的脂肪族杂环基。该4员至6员的脂肪族杂环基任选具有1至3个独立选自下述C组的取代基,
B组:卤素原子、羟基和C1~C6烷氧基。
C组:卤素原子、羟基、C1~C6烷基、C1~C6烷氧基和二C1~C6烷基氨基。
(1)自化合物2a向化合物4a的转化
在适当碱(例如氢化钠、三乙基胺、N,N-二(丙烷-2-基)乙基胺、N-甲基吗啉、4-二甲基氨基吡啶或它们的混合物)的存在下、在对反应没有不利影响的适当溶剂(例如苯、甲苯、乙醚、二氯甲烷、四氢呋喃或N,N-二甲基甲酰胺或这些混合溶剂)中、于-30℃至反应中使用的溶剂的沸点(优选0℃至100℃),自化合物2a至化合物4a的转化通过衍生自羧酸化合物2a的酰卤或羧酸活化酯的反应进行。可以使用催化量或者过量的碱。反应时间优选为10分钟至72小时,更优选30分钟至24小时。
(2)自化合物4a向化合物1a的转化
自化合物4a至化合物1a的转化根据R39的种类而采用不同的脱保护反应条件。当R39为甲基、乙基、苯甲基等时,在对反应不会产生不利影响的适当溶剂(其示例包括甲醇、乙醇、水、四氢呋喃、二烷等或它们的混合溶剂,优选以与水以任意比率可混合的有机溶剂)中,于-30℃至反应使用的溶剂的沸点,优选于室温至100℃,通过采用适当碱(例如氢氧化钠、氢氧化钾、氢氧化锂或叔丁醇钾)进行处理而实施所述转化。反应时间优选10分钟至72小时,更优选30分钟至24小时。
当R39为叔丁基等时,在对反应不会产生不利影响的适当溶剂(例如二氯甲烷、氯仿、甲醇、四氢呋喃、1,4-二烷或它们的混合溶剂)中,于-30℃至反应使用的溶剂的沸点,优选-20℃至室温,通过采用例如三氟乙酸、盐酸或甲酸处理进行所述转化。反应时间优选10分钟至72小时,更优选30分钟至24小时。
用于生产的原料化合物2a及3a可依据参考实施例记载的方法进行合成。
[生产方法2]
在式(1)所述化合物中,以下所述化合物1b可以通过例如下述反应流程生产:
式20
其中,R1、R31、R32、R33、R38、W、X、Y及Z如上文所定义。其中,虚线表示单键或双键。R39表示羧基的保护基。
R25表示环内具有1个氮原子的4员至6员的脂肪族杂环基。
该4员至6员的脂肪族杂环通过环上的氮原子与R26连接。该4员至6员的脂肪族杂环任选具有1至3个独立选自下述C组的取代基,桥接结构任选在杂环内连接,或者一个C3~C6环烷基环任选通过螺键连接到杂环环上。
R26表示氨基的保护基,示例包括2-硝基苯基磺酰基。
R27表示-C(=O)R23或-SO2R23。
R23表示C2~C6烯基或C2~C6炔基。
C组:卤素原子、羟基、氰基、任选具有1至3个独立选自下述D组的取代基的C1~C6烷基、C1~C6烷氧基和-NR21R22。
其中,R21及R22分别独立表示氢原子、C1~C6烷基或-C(=O)R23,R23表示C2~C6烯基或C2~C6炔基。
D组:氨基、C1~C6烷氧基、二C1~C6烷基氨基、氧代和C3~C6环烷基。
(1)自化合物2b向化合物4b的转化
自化合物2a向化合物4b的转化可通过与上述[生产方法1]的(1)所述方法相似的通用偶合反应进行。
(2)自化合物4b向化合物5b的转化
当R26为2-硝基苯基磺酰基等时,在对反应不会产生不利影响的适当溶剂(例如乙腈、N,N-二甲基甲酰胺等或它们的混合溶剂)中,于-30℃至反应使用的溶剂的沸点,优选于-20℃至室温,自化合物4b向化合物5b的转化可以通过采用适当的碱(例如碳酸钾或碳酸铯)和硫醇衍生物(例如苯硫醇)进行处理而实施。反应时间优选10分钟至72小时,优选30分钟至24小时。
(3)自化合物5b向化合物6b的转化
在对反应不会产生不利影响的适当溶剂(例如二氯甲烷、氯仿、四氢呋喃、乙酸乙酯、甲苯或它们的混合溶剂)中,在适当碱(例如,有机碱,如三乙基胺、N,N-二(丙烷-2-基)乙基胺、4-二甲基氨基吡啶、N-甲基吗啉、吡啶、2,6-卢剔啶、二氮杂双环[5.4.0]十一碳-7-烯;或无机碱,如碳酸钾、碳酸钠或碳酸氢钠)存在下,化合物5b向化合物6b的转化通过化合物5b与酰卤或磺酰卤的反应进行。反应温度的范围通常为-78℃至100℃或者至溶剂的沸点,优选为-10℃至室温附近。作为另外的方法,在对反应不会产生不利影响的适当溶剂(例如二氯甲烷、N,N-二甲基甲酰胺、四氢呋喃、乙酸乙酯或它们的混合溶剂)中,在适当的缩合剂(例如N,N’-二环己基碳二亚胺或1-乙基-3-(3-二甲基氨基丙基)碳二亚胺)存在下,本化合物6b可以通过化合物5b与羧酸或磺酸的反应而获得。反应温度范围通常为-78℃至100℃或者至溶剂的沸点,优选0℃至50℃。如果必要,可以向其中加入碱,例如三乙基胺、N,N-二(丙烷-2-基)乙基胺、N-甲基吗啉或4-二甲基氨基吡啶。此外,也可以向其中加入反应促进剂,例如1-羟基苯并三唑或N-羟基琥珀酸酰亚胺。
(4)自化合物6b向化合物1b的转化
自化合物6b向化合物1b的转化可通过与上述[生产方法1]的(2)所述方法相似的通用脱保护反应方法进行。
[生产方法3]
当R26为叔丁氧基羰基并且R39为[生产方法2]中的叔丁基时,化合物1b也可以通过下述制备方法合成:
式21
(1)自化合物4b向化合物7b的转化
在对反应不会产生不利影响的适当溶剂(例如二氯甲烷、氯仿、甲醇、四氢呋喃、1,4-二烷或它们的混合溶剂)中,于-30℃至反应使用的溶剂的沸点,优选于0℃至室温,自化合物4b向化合物7b的转化可以通过采用例如三氟乙酸、盐酸或甲酸处理而实施。反应时间优选10分钟至72小时,更优选30分钟至24小时。
(2)自化合物7b向化合物1b的转化
自化合物7b向化合物1b的转化可通过Schotten-Baumann反应而实施。具体地讲,使用对反应不会产生不利影响的适当溶剂(例如二氯甲烷或氯仿)和碱(例如碳酸氢钠或氢氧化钠)的水溶液,在双层系统条件下,通过采用酰卤或磺酰卤的反应而实施所述转化。反应温度优选为0℃至室温下,反应时间优选10分钟至72小时,更优选30分钟至24小时。
[生产方法4]
在式(1)所述化合物中,所述化合物1c可以通过例如下述反应流程生产:
式22
其中,R1、R31、R32、R33、R38、W、X、Y及Z如上文所定义。其中,虚线表示单键或双键。R39表示羧基的保护基。
R41和R42分别独立表示氢原子或C1~C6烷基,或者R41与R42可以任选一起形成环烷基环或环内具有氧原子的脂肪族杂环。
(1)自化合物2c向化合物4c的转化
在对反应不会产生不利影响的适当溶剂(例如N,N-二甲基甲酰胺、丙酮或它们的混合溶剂)中,在适当碱(例如碳酸钾、碳酸铯或它们的混合物)存在下,于室温至反应使用的溶剂的沸点,优选于50℃至100℃,自化合物2c向化合物4c的转化可以通过采用烷基卤化物的反应而实施。可使用过量的碱。反应时间优选1小时至72小时,更优选8小时至24小时。
(2)自化合物4c向化合物5c的转化
自化合物4c向化合物5c的转化可通过与上述[生产方法1]的(2)所述方法相似的通用脱保护反应方法进行。
(3)自化合物5c向化合物6c的转化
自化合物5c向化合物6c的转化可通过与上述[生产方法1]的(1)所述方法相似的通用酰胺化反应方法进行。
(4)自化合物6c向化合物1c的转化
自化合物6c向化合物1c的转化可通过与上述[生产方法1]的(2)所述方法相似的通用脱保护反应方法进行。
用于生产的原料化合物2c可依据参考实施例记载的方法合成。
[生产方法5]
在式(1)所述化合物中,以下所述化合物1c可以通过例如下述反应流程生产:
式23
其中,R1、R31、R32、R33、R38、W、X、Y及Z如上文所定义。其中,虚线表示单键或双键。R39表示羧基的保护基。
R41和R42分别独立表示氢原子或C1~C6烷基,或者R41与R42可以任选一起形成环烷基环或环内具有氧原子的脂肪族杂环。
(1)自化合物2d向化合物7c的转化
自化合物2d向化合物7c的转化可通过与上述[生产方法1]的(1)所述方法相似的通用酰胺化反应方法进行。
(2)自化合物7c向化合物6c的转化
在对反应不会产生不利影响的适当溶剂(例如N,N-二甲基甲酰胺、丙酮或它们的混合溶剂)中,于-30℃至反应使用的溶剂的沸点,优选于0℃至100℃,自化合物7c向化合物6c的转化可以通过相应的胺的反应进行。在该反应中,也可以使用过量的适当的碱(例如三乙基胺、N,N-二(丙烷-2-基)乙基胺、N-甲基吗啉、碳酸钾或它们的混合物)。反应时间优选10分钟至72小时,更优选30分钟至24小时。
(3)自化合物6c向化合物1c的转化
自化合物6c向化合物1c的转化可通过与上述[生产方法1]的(2)所述方法相似的通用脱保护反应方法进行。
用于生产的原料化合物2d可依据参考实施例记载的方法合成。
[生产方法6]
在式(1)所述化合物中,以下所述化合物1d可以通过例如下述反应流程生产:
式24
其中,R1、R31、R32、R33、R38、W、X、Y及Z如上文所定义。其中,虚线表示单键或双键。R39表示羧基的保护基,其示例包括叔丁基。
R51和R52分别独立表示氢原子或C1~C6烷基,或者R51与R52任选一起形成环烷基环。
R26表示氨基的保护基,其示例包括叔丁氧基羰基。
R27表示-C(=O)R23或-SO2R23。
(1)自化合物7c向化合物6d的转化
自化合物7c向化合物6d的转化可通过与上述[生产方法5]的(2)所述方法相似的通用取代反应方法进行。
(2)自化合物6d向化合物8d的转化
自化合物6d向化合物8d的转化可通过与上述[生产方法3]的(1)所述方法相似的通用脱保护反应方法进行。
(3)自化合物8d向化合物1d的转化
自化合物8d向化合物1d的转化可通过与上述[生产方法3]的(2)所述方法相似的酰化反应方法进行。
[生产方法7]
在式(1)所述化合物中,以下所述化合物1e可以通过例如下述反应流程生产:
式25
其中,R1、R31、R32、R33、R35、R36、Y及Z如上文所定义。R39表示羧基的保护基。
R28表示任选具有1至3个独立选自下述B组的取代基的C1~C6烷基、任选具有1至3个独立选自下述C组的取代基的C3~C6环烷基或环内具有1个氧原子的4员~6员的脂肪族杂环基,该4员~6员脂肪族杂环基可任选具有1至3个独立选自下述C组的取代基。
B组:卤素原子、羟基和C1~C6烷氧基。
C组:卤素原子、羟基、C1~C6烷基、C1~C6烷氧基和二C1~C6烷基氨基。
(1)自化合物2a向化合物4e的转化
自化合物2a向化合物4e的转化可通过Friedel-Crafts反应实施。具体地讲,在对反应无影响的适当溶剂(例如二氯甲烷)中,可以通过使用制备自化合物2a、化合物3b和金属氯化物(例如无水氯化铝或无水氯化锌)的酰氯而进行该转化。反应温度优选为-20℃至反应使用的溶剂的沸点,更优选为0℃至室温。优选所使用的金属氯化物为1至过量摩尔当量。
(2)自化合物4e向化合物5e的转化
自化合物4e向化合物5e的转化可通过Heck反应而实施。具体地讲,在对反应不会产生不利影响的适当溶剂(例如N,N-二甲基甲酰胺、四氢呋喃、甲苯、1,4-二烷或水或它们的混合溶剂)中,在相应的丙烯酸酯和适当的过渡金属催化剂(例如钯化合物)存在下,通过加入有机碱或无机碱(例如碳酸钠、碳酸钾、磷酸三钾或N,N-二(丙烷-2-基)乙基胺)和配体(例如三苯基膦)进行该转化。丙烯酸酯可获自商业,或者通过公知的方法生产。对于上述偶合反应,反应温度优选为0℃至300℃,更优选室温至200℃(最佳温度为80℃至100℃)。上述反应可以通过在密封管中或在微波照射下处理而进行。所述丙烯酸酯及碱各自优选使用相对于化合物4e的1至过量摩尔当量。更优选,丙烯酸酯的使用量为1至1.5摩尔当量,碱的使用量为1至5摩尔当量。反应时间优选1分钟至60小时,更优选5分钟至24小时。
(3)自化合物5e向化合物6e的转化
在对反应不会产生不利影响的适当溶剂(例如N,N-二甲基甲酰胺或四氢呋喃等)或它们的混合溶剂中,于-20℃至反应使用的溶剂的沸点,优选于0℃至室温,在适当碱(例如氢化钠)的存在下,自化合物5e向化合物6e的转化可以通过与烷基卤化物的反应而实施。可以使用过量的碱。反应时间优选为1小时至72小时,更优选8小时至24小时。
(4)自化合物6e向化合物1e的转化
自化合物6e向化合物1e的转化可以通过与上述[生产方法1]的(2)所述方法相似的通用脱保护反应方法进行。
[生产方法8]
在式(1)所述化合物中,以下所述化合物1f可以通过例如下述反应流程生产:
式26
其中,R1、R31、R32、R33、R35、R36、Y及Z如上文所定义。R39表示羧基的保护基。
R25表示环内具有1个氮原子的4员~6员的脂肪族杂环基,该4员至6员的脂肪族杂环通过在环上的氮原子与R26连接。该4员至6员的脂肪族杂环任选具有1至3个独立选自下述C组的取代基,并且
桥接结构任选在杂环内连接,或者一个C3~C6环烷基环任选通过螺连接合到杂环上。
R26表示氨基的保护基,其示例包括2-硝基苯基磺酰基。
R27表示-C(=O)R23或-SO2R23,
R23表示C2~C6烯基或C2~C6炔基,
C组:卤素原子、羟基、氰基、任选具有1至3个独立选自下述D组的取代基的C1~C6烷基、C1~C6烷氧基和-NR21R22。
其中,R21和R22分别独立表示氢原子、C1~C6烷基或-C(=O)R23,R23表示C2~C6烯基或C2~C6炔基。
D组:氨基、C1~C6烷氧基、二C1~C6烷基氨基、氧代基和C3~C6环烷基。
(1)自化合物2b向化合物4f的转化
自化合物2b向化合物4f的转化可以通过与上述[生产方法7]的(1)所述方法相似的通用偶合反应方法进行。
(2)自化合物4f向化合物5f的转化
自化合物4f向化合物5f的转化可以通过与上述[生产方法7]的(2)所述方法相似的通用烷基化反应方法进行。
(3)自化合物5f向化合物6f的转化
自化合物5f向化合物6f的转化可以通过与上述[生产方法7]的(3)所述方法相似的通用烷基化反应方法进行。
(4)自化合物6f向化合物7f的转化
自化合物6f向化合物7f的转化为可以通过与上述[生产方法2]的(2)所述方法相似的通用脱保护反应方法进行。
(5)自化合物7f向化合物8f的转化
自化合物7f向化合物8f的转化可以通过与上述[生产方法2]的(3)所述方法相似的通用酰化反应方法进行。
(6)自化合物8f向化合物1f的转化
自化合物8f向化合物1f的转化可以通过与上述[生产方法1]的(2)所述方法相似的通用脱保护反应方法进行。
[实施例]
在下文中,参照参考实施例、实施例及试验实施例对本发明做更详细说明。但本发明的范围不应当受到这些实施例的限定。
参考实施例和实施例中柱色谱的洗脱采用薄层色谱(TLC)(薄层色谱)观察。在TLC观察中,使用采用Merck公司生产的硅胶60F254或硅胶60NH2F254S作为TLC板;柱色谱中,将作为洗脱溶剂的溶剂用作展开溶剂,在检测方法中采用UV检测器。硅胶柱的硅胶SK-85(230~400目)同样由默克公司生产,或者采用Fuji Silysia Chemical Ltd.生产的Chromatorex NH(200-350目)。除通用柱色谱法外,也可以使用Yamazen Corp.的自动纯化装置(YFLC-5404-FC)或者Biotage Japan Ltd.Biotage公司的自动纯化装置(HORIZON、SP1或者Isolera)。使用的洗脱溶剂为在各参考实施例及实施例中所指定的溶剂。参考实施例及实施例中所使用的缩写具有下文所定义的意义。
mg:毫克,g:克,μl:微升,ml:毫升,L:升,MHz:兆赫。
在下面的实施例中,核磁共振(下文中称为1H NMR:400MHz)光谱通过采用四甲基硅烷作为标准测定的化学位移值δ值(ppm)表示。分裂模式表示为s=单峰,d=双峰,t=三重峰,q=四重峰,m=多重峰和br=宽峰。
[参考实施例K-1]3-(3-甲基氧杂环丁烷-3-基)-3-氧代丙酸乙基酯
式27
[步骤1]
向3-甲基氧杂环丁烷-3-甲酸(4.78g)的四氢呋喃(80ml)溶液中加入1,1’-羰基二咪唑(7.35g),将混合物在室温下搅拌1小时。向反应液加入丙二酸单乙酯钾(7.01g)及氯化镁(3.92g),将混合物在60℃下搅拌1小时。冷却后,将反应液用乙酸乙酯稀释并过滤。向滤液中加入1N盐酸使其呈酸性,分离两层,将有机层用饱和盐水洗涤,经无水硫酸钠干燥。减压蒸发溶剂后得到标题化合物(5.98g)。
1H-NMR(CDCl3)δ:1.29(3H,t,J=7.3Hz),1.60(3H,s),3.62(2H,s),4.21(2H,q,J=7.3Hz),4.45(2H,d,J=6.7Hz),4.91(2H,d,J=6.7Hz)。
通过与参考实施例K-1的步骤1相同的方法,得到下述化合物。
表1
表2
表3
[参考实施例K-15]4-氟-4-甲基-3-氧代戊酸乙基酯
式28
[步骤1]
向氢化钠(55%油分散液,6.10g)的四氢呋喃(40ml)悬浮液中用20分钟滴加2-氟-2-甲基丙酸乙酯(12.5g)与乙酸乙酯(12.3g)的混合液滴加,将混合物在室温下搅拌3小时。向反应液中加入1N盐酸中和,随后用正己烷进行萃取。将萃取液用饱和盐水洗涤,经无水硫酸镁干燥。减压蒸除溶剂,干燥残留物。除去上清油状物,得到粗品标题化合物(16.60g),直接用于下一反应。
1H-NMR(CDCl3)δ:1.29(3H,t,J=7.3Hz),1.50(6H,d,J=21.2Hz),3.66(2H,d,J=3.6Hz),4.21(2H,q,J=7.3Hz)。
[参考实施例K-16]4,4-二氟-3-氧代戊酸叔丁基酯
式29
[步骤1]
于-78℃向二(丙烷-2-基)氨化锂(1.12M的四氢呋喃溶液,36ml)的四氢呋喃(50ml)溶液中加入乙酸叔丁酯(4.83ml),将混合物在与上述相同的温度下搅拌30分钟。向反应液中加入2,2-二氟丙酸乙基酯(3g),将混合物在与上述相同的温度下搅拌2小时。向反应液中加入1N盐酸,用正己烷进行萃取。将萃取液以1N盐酸、饱和盐水依次洗涤,经无水硫酸镁干燥。减压蒸除溶剂后,得到粗品标题化合物(5.10g),直接用于下一反应。
1H-NMR(CDCl3)δ:1.47(9H,s),1.74(3H,t,J=19.3Hz),3.62(2H,s)。
通过与参考实施例K-16步骤1相同的方法,得到下述化合物。
表4
[参考实施例K-18]3-[1-(甲氧基甲氧基甲基)环丁基]-3-氧代丙酸乙基酯
式30
[步骤1]1-(甲氧基甲氧基甲基)环丁烷羧酸乙基酯
向1-(羟基甲基)环丁烷羧酸乙基酯(2.50g)的二氯甲烷(30ml)溶液中加入N,N-二(丙烷-2-基)乙基胺(4.06ml)、氯甲基甲基醚(1.43ml),于室温下将混合物搅拌1小时。再向反应液中加入氯甲基甲基醚(0.595ml)和N,N-二(丙烷-2-基)乙基胺(1.62ml),将混合物再搅拌1小时。向反应液中加入水和二氯甲烷,分离为两层。将有机层用0.25N盐酸、饱和碳酸氢钠水的依次洗涤,经无水硫酸钠干燥。再减压蒸除溶剂后得到标题化合物(2.98g)。
1H-NMR(CDCl3)δ:1.27(3H,t,J=7.3Hz),1.89-2.05(4H,m),2.39-2.48(2H,m),3.35(3H,s),3.79(2H,s),4.18(2H,q,J=7.3Hz),4.63(2H,s)。
[步骤2]1-(甲氧基甲氧基甲基)环丁烷羧酸
向由上述步骤1得到的化合物(2.98g)的甲醇(20ml)-四氢呋喃(20ml)混合液中加入1N氢氧化钠水溶液(20ml),将混合物在45℃下搅拌1.5小时。冷却后向反应液加入1N盐酸使其呈弱酸性,用乙酸乙酯萃取。将萃取液用饱和盐水洗涤,经无水硫酸钠干燥。经减压蒸除溶剂后得到标题化合物(2.05g)。
1H-NMR(CDCl3)δ:1.93-2.07(4H,m),2.42-2.53(2H,m),3.37(3H,s),3.82(2H,s),4.66(2H,s)。
[步骤3]3-[1-(甲氧基甲氧基甲基)环丁基]-3-氧代丙酸乙基酯
使用在上述步骤2获得的化合物(2.05g),通过与参考实施例K-1的步骤1相同的方法,得到标题化合物(2.49g)。
1H-NMR(CDCl3)δ:1.28(3H,t,J=7.3Hz),1.81-2.03(4H,m),2.40-2.48(2H,m),3.36(3H,s),3.54(2H,s),3.81(2H,s),4.19(2H,q,J=7.3Hz),4.61(2H,s)。
[参考实施例K-19]3-[1-(甲氧基甲基)环丁基]-3-氧代丙酸乙基酯
式31
[步骤1]1-(甲氧基甲基)环丁烷羧酸乙基酯
向1-(羟基甲基)环丁烷羧酸乙基酯(0.690g)的二氯甲烷(25ml)溶液中加入42%四氟硼酸水溶液(0.793ml),随后以小份加入(三甲硅烷基)重氮甲烷(2.0M的正己烷溶液,4.36ml),于室温下将混合物搅拌15分钟。向反应液加入水和二氯甲烷,分离为两层,将有机层经无水硫酸钠干燥。经减压蒸除溶剂后得到标题化合物(0.720g)。
1H-NMR(CDCl3)δ:1.27(3H,t,J=7.3Hz),1.88-2.05(4H,m),2.36-2.45(2H,m),3.36(3H,s),3.63(2H,s),4.18(2H,q,J=7.3Hz)。
[步骤2]1-(甲氧基甲基)环丁烷羧酸
使用上述步骤1获得的化合物(720mg),通过与参考实施例K-18步骤2相同的方法,得到标题化合物(300mg)。
1H-NMR(CDCl3)δ:1.92-2.06(4H,m),2.40-2.51(2H,m),3.41(3H,s),3.66(2H,s)。
[步骤3]3-[1-(甲氧基甲基)环丁基]-3-氧代丙酸乙基酯
使用在上述步骤2获得的化合物(285mg),通过与参考实施例K-1的步骤1相同的方法,得到标题化合物(308mg)。
1H-NMR(CDCl3)δ:1.28(3H,t,J=7.3Hz),1.81-2.00(4H,m),2.38-2.44(2H,m),3.34(3H,s),3.51(2H,s),3.64(2H,s),4.19(2H,q,J=7.3Hz)。
[参考实施例K-20]3-(1-甲氧基环丙基)-3-氧代丙酸乙基酯
式32
[步骤1]1-甲氧基环丙烷羧酸甲基酯
于冰冷却下向1-羟基环丙烷羧酸甲基酯(3.2g)的四氢呋喃(40ml)溶液中加入氢化钠(55%油分散液,1.3g),将混合物在与上述相同的温度下搅拌15分钟。向反应液中加入甲基碘(2.3ml),于室温下将混合物搅拌过夜。向反应液中加入1N盐酸,随后用正己烷-乙酸乙酯混合液进行萃取。将萃取液用饱和盐水洗涤,经无水硫酸镁干燥。经减压蒸除溶剂后得到标题化合物(4.0g)。
1H-NMR(CDCl3)δ:1.13-1.32(4H,m),3,42(3H,s),3.75(3H,s)。
[步骤2]1-甲氧基环丙烷羧酸
使用在上述步骤1获得的化合物(4.0g),通过与参考实施例K-18步骤2相同的方法,得到标题化合物(2.0g)。
1H-NMR(CDCl3)δ:1.22-1.26(2H,m),1.36-1.40(2H,m),3.45(3H,s)。
[步骤3]3-(1-甲氧基环丙基)-3-氧代丙酸乙基酯
使用在上述步骤2获得的化合物(2.0g),通过与参考实施例K-1的步骤1相同的方法,得到标题化合物(3.1g)。
1H-NMR(CDCl3)δ:1.22-1.39(7H,m),3.38(3H,s),3.72(2H,s),4.21(2H,q,J=7.3Hz)。
通过与参考实施例K-20相同的方法,得到下述化合物。
表5
[参考实施例K-22]3-{1-[(2-硝基苯基)磺酰基]哌啶-4-基}-3-氧代丙酸乙基酯
式33
[步骤1]1-(2-硝基苯基)磺酰基哌啶-4-甲酸
在冰冷却下向哌啶-4-甲酸(30g)的水溶液(1000ml)中,分小份加入碳酸钠(73.9g)及2-硝基苯磺酰氯(61.8g),于室温下将混合物搅拌过夜。向反应液中加入乙酸乙酯,分离两层。于冰冷却下,加入浓盐酸使其呈酸性,用乙酸乙酯进行萃取。将萃取液用水和饱和盐水依次洗涤,经无水硫酸钠干燥。减压蒸除溶剂,向获得的残留物中加入正己烷,过滤收集固体,得到标题化合物(69g)。
1H-NMR(CDCl3)δ:1.78-1.90(2H,m),1.99-2.07(2H,m),2.45-2.52(1H,m),2.93-3.02(2H,m),3.76(2H,dt,J=12.7,3.6Hz),7.61-7.74(3H,m),8.00(1H,dd,J=7.3,1.8Hz)。
[步骤2]3-{1-[(2-硝基苯基)磺酰基]哌啶-4-基}-3-氧代丙酸乙基酯
使用在上述步骤1获得的化合物(10g),通过与参考实施例K-1的步骤1相同的方法,得到标题化合物(14g)。
1H-NMR(CDCl3)δ:1.26(3H,t,J=7.3Hz),1.68-1.79(2H,m),1.84-2.01(2H,m),2.57-2.66(1H,m),2.88-2.97(2H,m),3.48(2H,s),3.79-3.86(2H,m),4.19(2H,q,J=7.3Hz),7.60-7.74(3H,m),7.98-8.02(1H,m)。
通过与参考实施例K-22相同的方法,得到下述化合物。
表6
[参考实施例K-25]4-(3-乙氧基-3-氧代丙酰基)-4-(甲氧基甲基)哌啶-1-甲酸叔丁基酯
式34
[步骤1]4-(甲氧基甲基)哌啶-1,4-二羧酸1-叔丁基4-乙基酯
在氮环境下,于-78℃向哌啶-1,4-二羧酸1-叔丁基4-乙基酯(1.00g)的四氢呋喃(20ml)溶液中加入二(丙烷-2-基)氨化锂(1.09M的四氢呋喃溶液,5.35ml),将混合物在与上述相同的温度下搅拌1小时。再向反应液中加入氯甲基甲基醚(0.73ml),将混合物逐渐升温至室温,于室温下搅拌过夜。向反应液中加入氯化铵水溶液(20ml),分离两层。将水层用乙酸乙酯进行萃取,合并有机层,用水和饱和盐水依次洗涤,经无水硫酸钠干燥。减压蒸除溶剂,将获得的残留物经硅胶柱色谱(正己烷/乙酸乙酯)进行纯化,得到标题化合物(917mg)。
1H-NMR(CDCl3)δ:1.27(3H,t,J=7.0Hz),1.38-1.48(11H,m),2.04-2.12(2H,m),2.85-3.06(2H,brm),3.30(3H,s),3.38(2H,brs),3.75-3.95(2H,brm),4.20(2H,q,J=7.1Hz)。
[步骤2]1-叔丁氧基羰基-4-(甲氧基甲基)哌啶-4-甲酸
使用在上述步骤1获得的化合物(910mg),通过与参考实施例K-18步骤3相同的方法,得到标题化合物(852mg)。
1H-NMR(CDCl3)δ:1.39-1.50(11H,m),2.03-2.12(2H,m),2.99-3.14(2H,brm),3.36(3H,s),3.44(2H,brs),3.75-3.91(2H,brm)。
MS(m/z):272(M-H)-。
[步骤3]4-(3-乙氧基-3-氧代丙酰基)-4-(甲氧基甲基)哌啶-1-甲酸叔丁基酯
使用在上述步骤2获得的化合物(825mg),通过与参考实施例K-1的步骤1相同的方法,得到标题化合物(835mg)。
1H-NMR(CDCl3)δ:1.27(3H,t,J=7.3Hz),1.44(9H,s),1.46-1.57(2H,brm),2.03(2H,ddd,J=13.9,4.1,2.0Hz),3.08-3.24(2H,m),3.30(3H,s),3.40(2H,s),3.57(2H,s),3.58-3.72(2H,brm),4.19(2H,q,J=7.3Hz)。
[参考实施例K-26]4-(3-乙氧基-3-氧代丙酰基)-4-甲氧基哌啶-1-甲酸叔丁基酯
式35
[步骤1]4-羟基哌啶-1,4-二羧酸1-叔丁基4-乙基酯
向4-羟基哌啶-4-甲酸乙基酯盐酸盐(1.02g)的二氯甲烷(10ml)悬浮液中加入三乙基胺(1.69ml)和二碳酸二叔丁基酯(1.23ml),于室温下将混合物搅拌过夜。将反应液减压浓缩,将获得的残留物经硅胶柱色谱(正己烷/乙酸乙酯)进行纯化,得到标题化合物(1.35g)。
1H-NMR(CDCl3)δ:1.30(3H,t,J=7.3Hz),1.47(9H,s),1.53-1.62(2H,brm),1.95(2H,td,J=12.7,4.8Hz),3.05(1H,s),3.07-3.25(2H,brm),3.86-4.08(2H,brm),4.25(2H,q,J=7.3Hz)。
[步骤2]4-甲氧基哌啶-1,4-二羧酸1-叔丁基4-乙基酯
使用在上述步骤1获得的化合物(685mg),通过与参考实施例K-20步骤1相同的方法,得到标题化合物(483mg)。
1H-NMR(CDCl3)δ:1.30(3H,t,J=7.0Hz),1.46(9H,s),1.82-1.96(4H,brm),3.10-3.23(2H,brm),3.27(3H,s),3.69-3.85(2H,brm),4.23(2H,q,J=7.3Hz)。
[步骤3]1-(叔丁氧基羰基)-4-甲氧基哌啶-4-甲酸
使用在上述步骤2获得的化合物(483mg),通过与参考实施例K-18步骤3相同的方法,得到标题化合物(430mg)。
1H-NMR(CDCl3)δ:1.46(9H,s),1.85-1.99(4H,m),3.08-3.23(2H,brm),3.33(3H,s),3.75-3.92(2H,brm)。
[步骤4]4-(3-乙氧基-3-氧代丙酰基)-4-甲氧基哌啶-1-甲酸叔丁基酯
使用在上述步骤3获得的化合物(430mg),通过与参考实施例K-1的步骤1相同的方法,得到标题化合物(488mg)。
1H-NMR(CDCl3)δ:1.28(3H,t,J=7.3Hz),1.45(9H,s),1.71-1.95(4H,m),3.08-3.22(2H,m),3.23(3H,s),3.63(2H,s),3.82-3.84(2H,brm),4.20(2H,q,J=7.3Hz)。
[参考实施例K-27]3-{4-(甲氧基甲氧基)-1-[(2-硝基苯基)磺酰基]哌啶-4-基}-3-氧代丙酸乙基酯
式36
[步骤1]4-羟基哌啶-4-甲酸乙基酯盐酸盐
在冰冷却下,向由参考实施例K-26的步骤1获得的化合物(2.78g)的乙醇溶液(30ml)中,加入4N盐酸-1,4-二烷溶液(10ml),于室温下将混合物搅拌过夜。将反应液在减压浓缩后得到标题化合物(2.12g)。
1H-NMR(DMSO-d6)δ:1.22(3H,t,J=7.3Hz),1.77-1.80(2H,m),2.02(2H,ddd,J=13.1,4.0,2.0Hz),3.02-3.05(2H,m),3.14-3.17(2H,m),4.14(2H,q,J=7.1Hz),5.86(1H,brs),8.51(1H,brs),8.67(1H,brs)。
[步骤2]4-羟基-1-[(2-硝基苯基)磺酰基]哌啶-4-甲酸乙基酯
于冰冷却下,向上述步骤1获得的化合物(2.12g)与三乙基胺(4.24ml)的二氯甲烷溶液(35ml)中,加入2-硝基苯磺酰氯(2.47g),于室温下将混合物搅拌过夜。将反应液用二氯甲烷稀释,加入水。分离两层,将有机层用水和饱和盐水依次洗涤,经无水硫酸钠干燥。减压蒸除溶剂,将获得的残留物经硅胶柱色谱(正己烷/乙酸乙酯)进行纯化,得到标题化合物(3.38g)。
1H-NMR(CDCl3)δ:1.31(3H,t,J=7.3Hz),1.68-1.71(2H,m),2.15(2H,ddd,J=13.1,4.6,2.3Hz),3.11(1H,s),3.16(2H,ddd,J=12.7,2.4,1.2Hz),3.78-3.81(2H,m),4.26(2H,q,J=7.1Hz),7.59-7.65(1H,m),7.67-7.73(2H,m),7.98(1H,dd,J=7.3,1.8Hz)。
MS(m/z):359(M+H)+。
[步骤3]4-(甲氧基甲氧基)-1-[(2-硝基苯基)磺酰基]哌啶-4-甲酸乙基酯
在冰冷却下,向在上述步骤2获得的化合物(1.51g)与氯甲基甲基醚(0.64ml)的N,N-二甲基甲酰胺溶液(16.9ml)中,加入氢化钠(55%的油分散液,369mg),于室温下将混合物搅拌30分钟。将反应液用乙酸乙酯稀释后,向反应液中加入冰,分离为两层。将有机层用水和饱和盐水依次洗涤,经无水硫酸钠干燥。减压蒸除溶剂,将获得的残留物经硅胶柱色谱(正己烷/乙酸乙酯)进行纯化,得到标题化合物(1.12g)。
1H-NMR(CDCl3)δ:1.28(3H,t,J=7.0Hz),2.01-2.13(4H,m),3.21-3.27(2H,m),3.34(3H,s),3.60(2H,ddd,J=8.6,4.2,2.1Hz),4.19(2H,q,J=7.1Hz),4.70(2H,s),7.61-7.74(3H,m),7.99-8.02(1H,m)。
[步骤4]4-(甲氧基甲氧基)-1-[(2-硝基苯基)磺酰基]哌啶-4-甲酸
使用在上述步骤3获得的化合物(1.12g),通过与参考实施例K-18步骤3相同的方法,得到标题化合物(1.07g)。
1H-NMR(CDCl3)δ:2.07-2.17(4H,m),3.21-3.28(2H,m),3.38(3H,s),3.64(2H,ddd,J=8.5,4.0,2.0Hz),4.75(2H,s),7.62-7.64(1H,m),7.67-7.74(2H,m),8.00-8.02(1H,m)。
[步骤5]3-{4-(甲氧基甲氧基)-1-[(2-硝基苯基)磺酰基]哌啶-4-基}-3-氧代丙酸乙基酯
使用在上述步骤4获得的化合物(1.07g),通过与参考实施例K-1的步骤1相同的方法,得到标题化合物(958mg)。
1H-NMR(CDCl3)δ:1.26(3H,t,J=7.3Hz),1.91-2.02(4H,m),3.23-3.30(2H,m),3.35(3H,s),3.61-3.64(4H,m),4.18(2H,q,J=7.3Hz),4.61(2H,s),7.62-7.64(1H,m),7.69-7.72(2H,m),7.99-8.02(1H,m)。
MS(m/z):443(M-H)-。
[参考实施例K-28]3-{4-(叔丁氧基羰基氨基)-1-[(2-硝基苯基)磺酰基]哌啶-4-基}-3-氧代丙酸乙基酯
式37
[步骤1]4-(叔丁氧基羰基氨基)-1-[(2-硝基苯基)磺酰基]哌啶-4-甲酸甲基酯
在冰冷却下,向4-(叔丁氧基羰基氨基)哌啶-4-甲酸甲基酯(1.06g)的四氢呋喃(11ml)及水(11ml)混合液中,加入碳酸钠(1.30g)和2-硝基苯磺酰氯(1.09g),于室温下将混合物搅拌过夜。将反应液减压浓缩,获得的残留物用乙酸乙酯稀释,用水和饱和盐水依次洗涤,经无水硫酸钠干燥。减压蒸除溶剂,将获得的残留物经硅胶柱色谱(正己烷/乙酸乙酯)进行纯化,得到标题化合物(1.66g)。
1H-NMR(CDCl3)δ:1.42(9H,s),2.01-2.09(2H,m),2.17-2.25(2H,m),3.17-3.26(2H,m),3.62(2H,td,J=8.9,4.2Hz),3.73(3H,s),4.64(1H,brs),7.63(1H,dd,J=7.3,1.8Hz),7.67-7.75(2H,m),8.00(1H,dd,J=7.0,2.7Hz)。
[步骤2]4-(叔丁氧基羰基氨基)-1-[(2-硝基苯基)磺酰基]哌啶-4-甲酸
使用在上述步骤1获得的化合物(1.66g),通过与参考实施例K-18步骤3相同的方法,得到标题化合物(1.46g)。
1H-NMR(CDCl3)δ:1.43(9H,s),2.01-2.12(2H,m),2.19-2.29(2H,m),3.25(2H,t,J=11.2Hz),3.57-3.67(2H,m),4.80(1H,brs),7.62-7.77(3H,m),7.97-8.04(1H,m)。
[步骤3]3-{4-(叔丁氧基羰基氨基)-1-[(2-硝基苯基)磺酰基]哌啶-4-基}-3-氧代丙酸乙基酯
使用在上述步骤2获得的化合物(1.46g),通过与参考实施例K-1的步骤1相同的方法,得到标题化合物(1.23g)。
1H-NMR(CDCl3)δ:1.26(3H,t,J=7.3Hz),1.43(9H,s),1.85-1.97(2H,brm),2.16-2.32(2H,m),3.27-3.38(2H,m),3.51-3.61(2H,m),3.59(2H,s),4.17(2H,q,J=7.3Hz),4.83(1H,brs),7.62-7.76(3H,m),7.99(1H,dd,J=7.9,1.8Hz)。
[参考实施例X-1]3-(2,6-二氯苯基)-5-(丙烷-2-基)-1,2-唑-4-甲酸
式38
[步骤1](1E)-2,6-二氯苯甲醛肟
将2,6-二氯苯甲醛(1.24g)悬浮于水(20ml)中,向该悬浮液中加入羟基胺盐酸盐(652mg)和碳酸钠(488mg),将混合物加热至回流2小时。冷却后,将反应液用二氯甲烷萃取,将萃取液经无水硫酸钠干燥。经减压蒸除溶剂后得到标题化合物(1.31g)。
1H-NMR(CDCl3)δ:7.24(1H,dd,J=8.6,7.4Hz),7.37(2H,d,J=8.0Hz),7.88(1H,brs),8.38(1H,s)。
[步骤2](1Z)-2,6-二氯-N-羟基苯并氢肟基氯
(Benzenecarboximidoyl chloride)
在于水浴冷却下,向在上述步骤1获得的化合物(1.00g)的N,N-二甲基甲酰胺(13ml)溶液中加入N-氯琥珀酸酰亚胺(737mg),于室温下将混合物搅拌过夜。向反应液中加入水,用乙醚进行萃取。将萃取液用饱和盐水洗涤,经无水硫酸钠干燥。经减压蒸除溶剂后得到标题化合物(1.28g),无须进行纯化下使用于下个反应。
1H-NMR(CDCl3)δ:7.27-7.41(3H,m),9.64-9.94(1H,m)。
[步骤3]3-(2,6-二氯苯基)-5-(丙烷-2-基)-1,2-唑-4-甲酸甲基酯
在冰冷却下,向4-甲基-3-氧代戊酸甲基酯(0.79ml)的四氢呋喃(6ml)溶液中,滴加甲醇钠(28%甲醇溶液,1.07ml)的甲醇(10ml)溶液,于室温下将混合物搅拌1小时。在冰冷却下向反应液中滴加上述步骤2获得的化合物(1.28g)的四氢呋喃(3ml)溶液,于室温下将混合物搅拌过夜。将反应液减压浓缩,向获得的残留物中加入水,用乙醚进行萃取。将萃取液用饱和盐水洗涤,经无水硫酸钠干燥。经减压蒸除溶剂后得到标题化合物(1.34g)。
1H-NMR(CDCl3)δ:1.44(6H,d,J=7.3Hz),3.67(3H,s),3.83-3.90(1H,m),7.29-7.44(3H,m)。
[步骤4]3-(2,6-二氯苯基)-5-(丙烷-2-基)-1,2-唑-4-甲酸
向上述步骤3获得的化合物(1.34g)的甲醇(10ml)溶液中加入1N氢氧化钠水溶液(10ml),将混合物在55℃搅拌9小时。将反应液减压浓缩,向获得的残留物中加入乙酸乙酯,分离两层。在冰冷却下将水层使用1N盐酸使其呈酸性,用乙酸乙酯进行萃取。将萃取液经无水硫酸钠干燥后,经减压蒸除溶剂后得到标题化合物(1.11g)。
1H-NMR(CDCl3)δ:1.43(6H,d,J=6.7Hz),3.85-3.92(1H,m),7.30-7.43(3H,m)。
使用市售的酯或者参考实施例中记载的酯,通过与参考实施例X-1相同的方法,得到下述化合物。
表7
[表8]
[表9]
[表10]
[表11]
[表12]
[表13]
[表14]
[表15]
[表16]
[表17]
[表18]
[表19]
[参考实施例X-52]5-(1,1-二氟乙基)-3-(2,4,6-三氯苯基)-1,2-唑-4-甲酸
式39
[步骤1]5-(1,1-二氟乙基)-3-(2,4,6-三氯苯基)-1,2-唑-4-甲酸叔丁基酯
向(1E)-2,4,6-三氯苯甲醛肟(1.90g)的N,N-二甲基甲酰胺(30ml)溶液中加入N-氯琥珀酸酰亚胺(1.19g),于室温下将混合物搅拌45分钟。向反应液中加入参考实施例K-16的步骤1获得的化合物(3.52g)和三乙基胺(1.88g),于室温下将混合物搅拌过夜。将反应液用乙酸乙酯稀释,用水和饱和盐水依次洗涤,经无水硫酸镁干燥。减压蒸除溶剂,将获得的残留物经硅胶柱色谱(正己烷/乙酸乙酯)进行纯化,得到标题化合物(3.00g)。
1H-NMR(CDCl3)δ:1.33(9H,s),2.20(3H,t,J=18.7Hz),7.47(2H,s)。
[步骤2]5-(1,1-二氟乙基)-3-(2,4,6-三氯苯基)-1,2-唑-4-甲酸
将上述步骤1获得的化合物(3.00g)、三氟乙酸(5ml)和二氯甲烷(15ml)的混合物于室温下搅拌3小时。将反应液用氯仿稀释,用水和饱和盐水依次洗涤,经无水硫酸镁干燥。减压蒸除溶剂,获得的残留物用正己烷洗涤,得到标题化合物(2.07g)。
1H-NMR(CDCl3)δ:2.21(3H,t,J=18.4Hz),7.46(2H,s)。
使用参考实施例记载的酯,通过与参考实施例X-52相同的方法,得到下述化合物。
[表20]
[参考实施例X-54]5-(6-氯吡啶-3-基)-3-(2,4,6-三氯苯基)-1,2-唑-4-甲酸
式40
[步骤1]5-(6-氯吡啶-3-基)-3-(2,4,6-三氯苯基)-1,2-唑-4-甲酸乙基酯
向(1E)-2,4,6-三氯-苯甲醛肟(1.00g)的N,N-二甲基甲酰胺(18ml)溶液中加入N-氯琥珀酸酰亚胺(625mg),于室温下将混合物搅拌过夜。将反应液倾至水中,用乙醚进行萃取。将萃取液用水和饱和盐水依次洗涤,经无水硫酸钠干燥。减压蒸除溶剂,将获得的残留物溶于N,N-二甲基甲酰胺(18ml)。向该溶液中依次加入参考实施例K-13获得的化合物(1.52g)与三乙基胺(1.55ml),于室温下将混合物搅拌6小时。将反应液倾至乙酸乙酯、正己烷和水的混合液中,分离两层。将有机层用水和饱和盐水依次洗涤,经无水硫酸钠干燥。减压蒸除溶剂,将获得的残留物经硅胶柱色谱(正己烷/乙酸乙酯)进行纯化,得到标题化合物(1.08g)。
1H-NMR(CDCl3)δ:1.03(3H,t,J=7.0Hz),4.15(2H,q,J=7.0Hz),7.48(2H,s),7.53(1H,d,J=8.5Hz),8.48(1H,dd,J=8.5,2.4Hz),9.13(1H,d,J=2.4Hz)。
MS(m/z):431(M+H)+。
[步骤2]5-(6-氯吡啶-3-基)-3-(2,4,6-三氯苯基)-1,2-唑-4-甲酸
向上述步骤1获得的化合物(570mg)的四氢呋喃(5ml)-甲醇(5ml)混合液中加入1N氢氧化钠水溶液(5ml),将混合物加热至回流1小时。冷却后,将反应液减压浓缩,于冰冷却下向获得的残留物中加入1N盐酸(7ml)。过滤收集沉积的固体,用水和正己烷依次洗涤,减压下加热干燥,得到粗品标题化合物(497mg),直接用于下一反应。
1H-NMR(CDCl3)δ:7.48(2H,s),7.54(1H,d,J=8.5Hz),8.40(1H,dd,J=7.9,2.4Hz),9.14(1H,s)。
MS(m/z):403(M+H)+。
[参考实施例X-55]5-{3-甲基-1-[(2-硝基苯基)磺酰基]氮杂环丁烷-3-基}-3-(2,4,6-三氯苯基)-1,2-唑-4-甲酸
式41
[步骤1]5-[1-(叔丁氧基羰基)-3-甲基氮杂环丁烷-3-基]-3-(2,4,6-三氯苯基)-1,2-唑-4-甲酸乙基酯
向(1E)-2,4,6-三氯-苯甲醛肟(536mg)的N,N-二甲基甲酰胺(6ml)溶液中加入N-氯琥珀酸酰亚胺(319mg),于室温下将混合物搅拌过夜。在冰冷却下向反应液中加入甲醇钠(28%甲醇溶液、0.48ml),然后向其中滴加通过将参考实施例K-10获得的化合物(681mg)的四氢呋喃(7ml)溶液和甲醇钠(28%甲醇溶液、0.48ml)的甲醇(5ml)溶液混合制备制的烯醇溶液。于室温下将混合物搅拌过夜,减压浓缩。将获得的残留物用乙酸乙酯稀释,用水和饱和盐水依次洗涤,经无水硫酸钠干燥。减压蒸除溶剂,将获得的残留物经硅胶柱色谱(正己烷/乙酸乙酯)进行纯化,得到标题化合物(0.80g)。
1H-NMR(CDCl3)δ:1.46(9H,s),1.80(3H,s),4.03(2H,d,J=9.1Hz),4.14(2H,q,J=7.1Hz),4.46(2H,d,J=9.1Hz),7.45(2H,s)。
[步骤2]5-(3-甲基氮杂环丁烷-3-基)-3-(2,4,6-三氯苯基)-1,2-唑-4-甲酸乙基酯盐酸盐
向上述步骤1获得的化合物(0.80g)中加入4N盐酸-1,4-二烷(8ml),于室温下将混合物搅拌1.5小时。将反应液减压浓缩,得到标题化合物,直接用于下一反应。
[步骤3]5-{3-甲基-1-[(2-硝基苯基)磺酰基]氮杂环丁烷-3-基}-3-(2,4,6-三氯苯基)-1,2-唑-4-甲酸乙基酯
在冰冷却下向上述步骤2获得的化合物的二氯甲烷(8ml)悬浮液中加入三乙基胺(0.91ml)及2-硝基苯磺酰氯(470mg),于室温下将混合物搅拌过夜。将反应液用二氯甲烷稀释,用水和饱和盐水依次洗涤,经无水硫酸钠干燥。减压蒸除溶剂,将获得的残留物经硅胶柱色谱(正己烷/乙酸乙酯)进行纯化,得到标题化合物(454mg)。
1H-NMR(CDCl3)δ:1.08(3H,t,J=7.0Hz),1.81(3H,s),4.08-4.21(4H,m),4.67(2H,d,J=8.5Hz),7.44(2H,s),7.69-7.75(3H,m),8.08(1H,dd,J=7.3,2.4Hz)。
[步骤4]5-{3-甲基-1-[(2-硝基苯基)磺酰基]氮杂环丁烷-3-基}-3-(2,4,6-三氯苯基)-1,2-唑-4-甲酸
使用上述步骤3获得的化合物(334mg),通过与参考实施例X-1步骤1相同的方法,得到标题化合物(312mg)。
1H-NMR(CDCl3)δ:1.83(3H,s),4.16(2H,d,J=8.5Hz),4.69(2H,d,J=8.5Hz),7.44(2H,s),7.70-7.76(3H,m),8.06-8.11(1H,m)。
使用参考实施例记载的酯,通过与参考实施例X-55相同的方法,得到下述化合物。
[表21]
[表22]
[参考实施例X-60]5-[1-(二甲基氨基)环丁基]-3-(2,4,6-三氯苯基)-1,2-唑-4-甲酸
式42
[步骤1]5-[1-(叔丁氧基羰基氨基)环丁基]-3-(2,4,6-三氯苯基)-1,2-唑-4-甲酸乙基酯
使用(1E)-2,4,6-三氯苯甲醛肟(79mg)和参考实施例K-8获得的化合物(100mg),通过与参考实施例X-55步骤1相同的方法,得到标题化合物(21mg)。
1H-NMR(CDCl3)δ:1.01(3H,t,J=7.3Hz),1.34(9H,s),1.93-2.04(1H,m),2.13-2.24(1H,m),2.48-2.55(2H,m),2.84-2.91(2H,m),4.10(2H,q,J=7.3Hz),5.79(1H,brs),7.44(1H,s)。
MS(m/z):489(M+H)+。
[步骤2]5-[1-(叔丁氧基羰基氨基)环丁基]-3-(2,4,6-三氯苯基)-1,2-唑-4-甲酸
使用在上述步骤1获得的化合物(2.50g),通过与参考实施例X-1步骤4相同的方法,得到标题化合物(2.45g)。
1H-NMR(DMSO-d6)δ:1.28(9H,brs),1.80-1.88(1H,m),2.00-2.11(1H,m),2.41-2.48(2H,m),2.70-2.77(2H,m),7.87(2H,s),13.01(1H,brs)。
[步骤3]5-(1-氨基环丁基)-3-(2,4,6-三氯苯基)-1,2-唑-4-甲酸盐酸盐
向上述步骤2获得的化合物(2.45g)的甲醇(20ml)悬浮液中加入4N盐酸的1,4-二烷溶液(15ml),于室温下将混合物搅拌1小时。再向反应液中加入4N盐酸的1,4-二烷溶液(5ml),进一步于室温下搅拌混合液1小时。将反应液减压浓缩,向获得的残留物中加入少量乙醚和正己烷,过滤收集产生的固体,得到标题化合物(1.55g)。
1H-NMR(DMSO-d6)δ:1.88-1.99(1H,m),2.21-2.32(1H,m),2.67-2.75(2H,m),2.81-2.90(2H,m),7.93(2H,s),9.20(3H,brs)。
[步骤4]5-[1-(二甲基氨基)环丁基]-3-(2,4,6-三氯苯基)-1,2-唑-4-甲酸
向上述步骤3获得的化合物(1.55g)的1,2-二氯乙烷(20ml)悬浮液中加入甲醛(37%水溶液,1.45ml)、N,N-二(丙烷-2-基)乙基胺(0.666ml)和三乙酰氧基硼氢化钠(4.13g),于室温下将混合物搅拌2小时。向反应液中加入水和二氯甲烷,分离两层,将水层用二氯甲烷进行萃取。合并有机层,经无水硫酸钠干燥。减压蒸除溶剂,向获得的残留物中加入正己烷,过滤收集产生的固体,得到标题化合物(1.25g)。
1H-NMR(DMSO-d6)δ:1.91-1.99(2H,m),2.42-2.52(2H,m),2.43(6H,s),2.71-2.80(2H,m),7.87(2H,s)。
MS(m/z):389(M+H)+。
使用参考实施例记载的酯,通过与参考实施例X-60相同的方法,得到下述化合物。
[表23]
[参考实施例X-62]5-(1-{甲基-[(2-硝基苯基)磺酰基]氨基}环丁基)-3-(2,4,6-三氯苯基)-1,2-唑-4-甲酸
式43
[步骤1]5-(1-氨基环丁基)-3-(2,4,6-三氯苯基)-1,2-唑-4-甲酸乙基酯盐酸盐
向在参考实施例X-60的步骤1获得的化合物(1.00g)的二氯甲烷(5ml)溶液中加入4N盐酸的1,4-二烷(10ml)溶液,于室温下将混合物搅拌1小时。将反应液减压浓缩,得到标题化合物,直接用于下一反应。
[步骤2]5-(1-{[(2-硝基苯基)磺酰基]氨基}环丁基)-3-(2,4,6-三氯苯基)-1,2-唑-4-甲酸乙基酯
向上述步骤1获得的化合物的二氯甲烷(20ml)悬浮液中加入三乙基胺(0.848ml)和2-硝基苯磺酰氯(0.452g),于室温下将混合物搅拌1小时。再向反应液中加入三乙基胺(0.283ml)和2-硝基苯磺酰氯(0.206g),进一步于室温下搅拌30分钟。向反应液加入水和二氯甲烷,分离两层,将有机层经无水硫酸钠干燥。减压蒸除溶剂,将获得的残留物经硅胶柱色谱(正己烷/乙酸乙酯)进行纯化,得到标题化合物(0.66g),直接用于下一反应。
[步骤3]5-[(1-{甲基-[(2-硝基苯基)磺酰基]氨基}环丁基)-3-(2,4,6-三氯苯基)-1,2-唑-4-甲酸乙基酯
向上述步骤2获得的化合物(53mg)的N,N-二甲基甲酰胺(3ml)溶液加入甲基碘(0.017ml)及氢化钠(55%的油分散液,6mg),于室温下将混合物搅拌15分。向反应液中加入水、乙酸乙酯,分离两层。将有机层用饱和盐水洗涤,经无水硫酸钠干燥。减压蒸除溶剂,将获得的残留物经硅胶柱色谱(正己烷/乙酸乙酯)进行纯化,得到标题化合物(31mg)。
1H-NMR(CDCl3)δ:0.95(3H,t,J=7.3Hz),1.71-1.83(1H,m),1.88-1.97(1H,m),2.83-2.93(2H,m),2.98-3.04(2H,m),3.11(3H,s),4.05(2H,q,J=7.3Hz),7.44(2H,s),7.57-7.71(3H,m),7.92-7.95(1H,m)。
[步骤4]5-(1-{甲基-[(2-硝基苯基)磺酰基]氨基}环丁基)-3-(2,4,6-三氯苯基)-1,2-唑-4-甲酸
使用在上述步骤3获得的化合物,通过与参考实施例X-1步骤4相同的方法,得到标题化合物,直接用于下一反应。
使用参考实施例记载的酯,通过与参考实施例X-60和X-62相同的方法,得到下述化合物。
[表24]
[参考实施例X-65]5-{1-[(叔丁氧基羰基)(丙-2-烯-1-基)氨基]环丁基}-3-(2,4,6-三氯苯基)-1,2-唑-4-甲酸
式44
[步骤1]5-{1-[(叔丁氧基羰基)(丙-2-烯-1-基)氨基]环丁基}-3-(2,4,6-三氯苯基)-1,2-唑-4-甲酸乙基酯
向氢化钠(55%的油分散液,0.260g)的N,N-二甲基甲酰胺(5ml)悬浮液中加入参考实施例X-60的步骤1获得的化合物(1.46g),于室温下将混合物搅拌5分钟。于冰冷却下向反应液中加入烯丙基溴(1.80g),于室温下将混合物搅拌1小时。在冰冷却下向反应液加入水和乙酸乙酯,分离两层。将水层用乙酸乙酯进行萃取,合并有机层,用饱和盐水洗涤,经无水硫酸钠干燥。减压蒸除溶剂,将获得的残留物经硅胶柱色谱(正己烷/乙酸乙酯)进行纯化,得到标题化合物(0.871g)。
1H-NMR(CDCl3)δ:0.96(3H,t,J=7.3Hz),1.35(9H,s),1.86-1.96(2H,m),2.66-2.74(2H,m),2.90-2.96(2H,m),4.05(2H,q,J=7.3Hz),4.16-4.23(2H,m),5.19-5.30(2H,m),5.94-6.05(1H,m),7.44(2H,s)。
MS(m/z):529(M+H)+。
[步骤2]5-{1-[(叔丁氧基羰基)(丙-2-烯-1-基)氨基]环丁基}-3-(2,4,6-三氯苯基)-1,2-唑-4-甲酸
使用在上述步骤1获得的化合物(0.871g),通过与参考实施例X-1步骤4相同的方法,得到标题化合物,直接用于下一反应。
1H-NMR(CDCl3)δ:1.40(9H,s),1.80-1.88(1H,m),1.90-2.00(1H,m),2.66-2.76(2H,m),2.89-2.99(2H,m),4.04-4.08(2H,m),5.21-5.27(2H,m),5.89-5.98(1H,m),7.43(2H,s)。
[参考实施例X-66]5-[1-(二甲基氨基)环丙基]-3-(2,4,6-三氯苯基)-1,2-唑-4-甲酸
式45
[步骤1]5-[1-(叔丁氧基羰基氨基)环丙基]-3-(2,4,6-三氯苯基)-1,2-唑-4-甲酸乙基酯
使用(1E)-2,4,6-三氯苯甲醛肟(2.42g)和参考实施例K-7获得的化合物(2.93g),通过与参考实施例X-55步骤1相同的方法,得到标题化合物(3.32g)。
1H-NMR(CDCl3)δ:1.03(3H,t,J=7.3Hz),1.39(9H,s),1.40-1.44(2H,m),1.61-1.66(2H,m),4.14(2H,q,J=7.3Hz),6.03(1H,brs),7.43(2H,s)。
MS(m/z):475(M+H)+。
[步骤2]5-(1-氨基环丙基)-3-(2,4,6-三氯苯基)-1,2-唑-4-甲酸乙基酯盐酸盐
使用在上述步骤1获得的化合物(3.32g),通过与参考实施例X-62的步骤1相同的方法,得到标题化合物(2.40g)。
1H-NMR(DMSO-d6)δ:1.02(3H,t,J=7.3Hz),1.68-1.72(4H,m),4.13(2H,q,J=7.3Hz),7.96(2H,s),9.14(3H,brs)。
[步骤3]5-[1-(二甲基氨基)环丙基]-3-(2,4,6-三氯苯基)-1,2-唑-4-甲酸乙基酯
使用在上述步骤2获得的化合物(1.00g),通过与参考实施例X-60的步骤4相同的方法,得到标题化合物(584mg)。
1H-NMR(CDCl3)δ:1.10(3H,t,J=7.3Hz),1.20-1.21(4H,m),2.38(6H,s),4.14(2H,q,J=7.3Hz),7.44(2H,s)。
[步骤4]5-[1-(二甲基氨基)环丙基]-3-(2,4,6-三氯苯基)-1,2-唑-4-甲酸
使用上述步骤3获得的化合物(584mg),通过与参考实施例X-1步骤4相同的方法,得到标题化合物,直接用于下一反应。
[参考实施例X-67]3-(2,6-二氯苯基)-5-(3-甲氧基氧杂环丁烷-3-基)-1,2-唑-4-甲酸
式46
[步骤1]3-(3-羟基氧杂环丁烷-3-基)丙-2-酸甲基酯
在-78℃下,向双(三甲硅烷基)氨化锂(1.09M的四氢呋喃溶液,9.2ml)溶液中滴加丙炔酸甲基酯(0.83ml)的四氢呋喃(10ml)溶液,将混合物在与上述相同的温度下搅拌30分钟。向反应液中加入氧杂环丁烷-3-酮(360mg),将混合物搅拌1.5小时后,倾至乙酸乙酯和饱和氯化铵水溶液中。剧烈搅拌,分离两层,将有机层用水和饱和盐水依次洗涤,然后经无水硫酸钠干燥。减压蒸除溶剂,将获得的残留物经硅胶柱色谱(正己烷/乙酸乙酯)进行纯化,得到标题化合物(152mg)。
1H-NMR(CDCl3)δ:2.76(1H,s),3.82(3H,s),4.73(2H,d,J=6.7Hz),4.88(2H,d,J=6.7Hz)。
[步骤2]3-(2,6-二氯苯基)-5-(3-羟基氧杂环丁烷-3-基)-1,2-唑-4-甲酸甲基酯
向参考实施例X-1的步骤1获得的化合物(190mg)的N,N-二甲基甲酰胺(2ml)溶液中加入N-氯琥珀酸酰亚胺(134mg),于室温下将混合物搅拌过夜。在冰冷却下,向反应液中加入三乙基胺(0.34ml)和上述步骤1获得的化合物(152mg)的N,N-二甲基甲酰胺(2ml)溶液,于室温下将混合物搅拌两天。将反应液用乙酸乙酯稀释,用水和饱和盐水依次洗涤,经无水硫酸钠干燥。减压蒸除溶剂,将获得的残留物经硅胶柱色谱(正己烷/乙酸乙酯)进行纯化,得到标题化合物(281mg)。
1H-NMR(CDCl3)δ:3.66(3H,s),4.97(2H,d,J=7.7Hz),5.15(2H,d,J=7.7Hz),5.87(1H,s),7.36-7.46(3H,m)。
[步骤3]3-(2,6-二氯苯基)-5-(3-甲氧基氧杂环丁烷-3-基)-1,2-唑-4-甲酸甲基酯
在冰冷却下,向上述步骤2获得的化合物(124mg)的N,N-二甲基甲酰胺(2ml)溶液中加入甲基碘(0.050ml)和氢化钠(55%的油分散液,22mg),于室温下将混合物搅拌20分钟。在冰冷却下,将反应液用乙酸乙酯稀释,用水和饱和盐水依次洗涤,经无水硫酸钠干燥。减压蒸除溶剂,将获得的残留物经硅胶柱色谱(正己烷/乙酸乙酯)进行纯化,得到标题化合物(93.3mg)。
1H-NMR(CDCl3)δ:3.23(3H,s),3.67(3H,s),4.98(2H,d,J=6.8Hz),5.18(2H,d,J=6.8Hz),7.34-7.50(3H,m)。
[步骤4]3-(2,6-二氯苯基)-5-(3-甲氧基氧杂环丁烷-3-基)-1,2-唑-4-甲酸
使用上述步骤3获得的化合物(73mg),通过与参考实施例X-1步骤4相同的方法,得到标题化合物(67mg)。
1H-NMR(CDCl3)δ:3.26(3H,s),4.97(2H,d,J=7.7Hz),5.18(2H,d,J=7.7Hz),7.36-7.46(3H,m)。
通过与参考实施例X-67相同的方法,得到下述化合物。
[表25]
[参考实施例X-70]5-(3-氟氧杂环丁烷-3-基)-3-(2,4,6-三氯苯基)-1,2-唑-4-甲酸
式47
[步骤1]3-(3-羟基氧杂环丁烷-3-基)丙-2-炔酸叔丁基酯
使用氧杂环丁烷-3-酮(3.0g),通过与参考实施例X-67的步骤1相同的方法,得到标题化合物(8.25g)。
1H-NMR(CDCl3)δ:1.51(9H,s),3.16(1H,s),4.72(2H,d,J=6.7Hz),4.89(2H,d,J=6.7Hz)。
[步骤2]5-(3-羟基氧杂环丁烷-3-基)-3-(2,4,6-三氯苯基)-1,2-唑-4-甲酸叔丁基酯
使用在上述步骤1获得的化合物(861mg),通过与参考实施例X-67的步骤2相同的方法,得到标题化合物(1.27g)。
1H-NMR(CDCl3)δ:1.25(9H,s),4.96(2H,d,J=7.3Hz),5.13(2H,d,J=7.3Hz),6.33(1H,s),7.47(2H,s)。
[步骤3]5-(3-氟氧杂环丁烷-3-基)-3-(2,4,6-三氯苯基)-1,2-唑-4-甲酸叔丁基酯
在-78℃下,向上述步骤2获得的化合物(87mg)的二氯甲烷(1ml)溶液中加入(二乙基氨基)三氟化硫(0.030ml),将混合物在与上述相同的温度下搅拌30分钟,然后在0℃搅拌1小时。将反应液用二氯甲烷稀释,用水和饱和盐水依次洗涤,经无水硫酸钠干燥。减压蒸除溶剂,将获得的残留物经硅胶柱色谱(正己烷/乙酸乙酯)进行纯化,得到标题化合物(66mg)。
1H-NMR(CDCl3)δ:1.31(9H,s),5.18(2H,dd,J=20.9,8.8Hz),5.26(2H,dd,J=22.4,8.5Hz),7.47(2H,s)。
[步骤4]5-(3-氟氧杂环丁烷-3-基)-3-(2,4,6-三氯苯基)-1,2-唑-4-甲酸
使用上述步骤3获得的化合物(66mg),通过与参考实施例X-52的步骤2相同的方法,得到标题化合物(57mg)。
1H-NMR(CDCl3)δ:5.18(2H,dd,J=22.1,9.4Hz),5.28(2H,dd,J=22.4,9.1Hz),7.47(2H,s)。
[参考实施例X-71]3-(2,6-二氯苯基)-5-[1-(甲氧基甲氧基)-1-甲基乙基]-1,2-唑-4-甲酸
式48
[步骤1]4-羟基-4-甲基戊-2-炔酸乙基酯
使用丙炔酸乙基酯(1.02ml),通过与参考实施例X-67的步骤1相同的方法,得到标题化合物(1.64g)。
1H-NMR(CDCl3)δ:1.32(3H,t,J=7.3Hz),1.57(6H,s),2.05(1H,s),4.24(2H,q,J=7.1Hz)。
[步骤2]3-(2,6-二氯苯基)-5-(1-羟基-1-甲基乙基)-1,2-唑-4-甲酸乙基酯
使用在上述步骤1获得的化合物(781mg),通过与参考实施例X-67的步骤2相同的方法,得到标题化合物(627mg)。
1H-NMR(CDCl3)δ:0.89(3H,t,J=7.3Hz),1.73(6H,s),4.10(2H,q,J=7.1Hz),5.96(1H,s),7.33-7.43(3H,m)。
[步骤3]3-(2,6-二氯苯基)-5-[1-(甲氧基甲氧基)-1-甲基乙基]-1,2-唑-4-甲酸乙基酯
在冰冷却下,向上述步骤2获得的化合物(304mg)的N,N-二甲基甲酰胺(3ml)溶液,加入氯甲基甲基醚(0.13ml)和氢化钠(55%的油分散液,77mg),于室温下将混合物搅拌过夜。将反应液用乙酸乙酯稀释,用水和饱和盐水依次洗涤,经无水硫酸钠干燥。减压蒸除溶剂,将获得的残留物经硅胶柱色谱(正己烷/乙酸乙酯)进行纯化,得到标题化合物(317mg)。
1H-NMR(CDCl3)δ:0.93(3H,t,J=7.0Hz),1.86(6H,s),3.36(3H,s),4.07(2H,q,J=7.3Hz),4.78(2H,s),7.31-7.45(3H,m)。
[步骤4]3-(2,6-二氯苯基)-5-[1-(甲氧基甲氧基)-1-甲基乙基]-1,2-唑-4-甲酸
使用上述步骤3获得的化合物(317mg),通过与参考实施例X-1步骤4相同的方法,得到标题化合物(296mg)。
1H-NMR(CDCl3)δ:1.89(6H,s),3.44(3H,s),4.96(2H,s),7.33-7.43(3H,m)。
[参考实施例X-72]3-(2,6-二氯苯基)-5-(2-甲基氧杂环丁烷-2-基)-1,2-唑-4-甲酸
式49
[步骤1]4-[叔丁基(二甲基)硅基]氧基丁烷-2-酮
在冰冷却下,向4-羟基丁烷-2-酮(881mg)的二氯甲烷(26ml)溶液中依次加入三乙基胺(4.2ml)、4-二甲基氨基吡啶(244mg)和叔丁基二甲基氯硅烷(1.81g),于室温下将混合物搅拌过夜。将反应液用二氯甲烷稀释,用水和饱和盐水依次洗涤,经无水硫酸钠干燥。减压蒸除溶剂,将获得的残留物经硅胶柱色谱(正己烷/乙酸乙酯)进行纯化,得到标题化合物(1.63g)。
1H-NMR(CDCl3)δ:0.05(6H,s),0.88(9H,s),2.18(3H,s),2.62(2H,t,J=6.0Hz),3.89(2H,t,J=6.3Hz)。
[步骤2]6-[叔丁基(二甲基)硅基]氧基-4-羟基-4-甲基己-2-炔酸乙基酯
使用在上述步骤1获得的化合物(809mg),通过与参考实施例X-67的步骤1相同的方法,得到标题化合物(1.09g)。
1H-NMR(CDCl3)δ:0.89(3H,t,J=7.3Hz),1.73(6H,s),4.10(2H,q,J=7.1Hz),5.96(1H,s),7.33-7.43(3H,m)。
[步骤3]5-[3-[叔丁基(二甲基)硅基]氧基-1-羟基-1-甲基丙基]-3-(2,6-二氯苯基)-1,2-唑-4-甲酸乙基酯
使用在上述步骤2获得的化合物(1.09g),通过与参考实施例X-67的步骤2相同的方法,得到标题化合物(850mg)。
1H-NMR(CDCl3)δ:0.04(3H,s),0.05(3H,s),0.88(3H,t,J=7.3Hz),0.89(9H,s),1.74(3H,s),2.22(1H,td,J=14.5,7.5Hz),2.32-2.40(1H,m),3.74(2H,t,J=7.0Hz),4.07(2H,q,J=7.1Hz),5.90(1H,s),7.32-7.46(3H,m)。
[步骤4]3-(2,6-二氯苯基)-5-(1,3-二羟基-1-甲基丙基)-1,2-唑-4-甲酸乙基酯
向上述步骤3获得的化合物(434mg)的甲醇(5ml)和四氢呋喃(5ml)混合液中加入吡啶鎓对-甲苯磺酸盐(223mg),于室温下将混合物搅拌过夜。将反应液减压浓缩,将获得的残留物经硅胶柱色谱(正己烷/乙酸乙酯)进行纯化,得到标题化合物(293mg)。
1H-NMR(CDCl3)δ:0.89(3H,t,J=7.3Hz),1.74(3H,s),2.17-2.25(1H,m),2.43-2.51(1H,m),2.70(1H,t,J=5.4Hz),3.76-3.85(1H,m),3.88-3.96(1H,m),4.10(2H,q,J=7.3Hz),6.47(1H,s),7.34-7.44(3H,m)。
[步骤5]3-(2,6-二氯苯基)-5-(2-甲基氧杂环丁烷-2-基)-1,2-唑-4-甲酸乙基酯
在冰冷却下,向上述步骤4获得的化合物(355mg)的二氯甲烷(5ml)溶液加入三乙基胺(0.18ml)及甲磺酰氯(0.090ml),将混合物在与上述相同的温度下搅拌15分钟。向反应液加入四氢呋喃(20ml)及叔丁醇钾(426mg),于室温下将混合物搅拌20分钟。在冰冷却下将反应液用乙酸乙酯稀释,用水和饱和盐水依次洗涤,经无水硫酸钠干燥。减压蒸除溶剂,将获得的残留物经硅胶柱色谱(正己烷/乙酸乙酯)进行纯化,得到标题化合物(55.2mg)。
1H-NMR(CDCl3)δ:0.99(3H,t,J=7.3Hz),1.96(3H,s),2.97(1H,dt,J=14.5,5.9Hz),3.22(1H,dt,J=14.5,5.7Hz),4.09(2H,q,J=7.3Hz),4.66-4.73(2H,m),7.32-7.43(3H,m)。
[步骤6]3-(2,6-二氯苯基)-5-(2-甲基氧杂环丁烷-2-基)-1,2-唑-4-甲酸
使用上述步骤5获得的化合物(95mg),通过与参考实施例X-1步骤4相同的方法,得到标题化合物(68mg)。
1H-NMR(CDCl3)δ:2.03(3H,s),3.05-3.14(1H,m),3.20(1H,ddd,J=13.8,7.1,4.7Hz),4.88(1H,dt,J=11.3,4.5Hz),4.92-5.00(1H,m),7.33-7.44(3H,m)。
[参考实施例X-73]5-[3-甲氧基-1-(2-硝基苯基)磺酰基氮杂环丁烷-3-基]-3-(2,4,6-三氯苯基)-1,2-唑-4-甲酸
式50
[步骤1]3-(3-乙氧基-3-氧代-1-丙炔基)-3-羟基氮杂环丁烷-1-甲酸叔丁基酯
使用3-氧代氮杂环丁烷-1-甲酸叔丁基酯(856mg),通过与参考实施例X-67的步骤1相同的方法,得到标题化合物(758mg)。
1H-NMR(CDCl3)δ:1.33(3H,t,J=7.0Hz),1.44(9H,s),2.87(1H,brs),4.05(2H,d,J=9.1Hz),4.23-4.30(4H,m)。
[步骤2]5-[1-(叔丁氧基羰基)-3-羟基氮杂环丁烷-3-基]-3-(2,4,6-三氯苯基)-1,2-唑-4-甲酸乙基酯
使用在上述步骤1获得的化合物(758mg),通过与参考实施例X-67的步骤2相同的方法,得到标题化合物(876mg)。
1H-NMR(CDCl3)δ:1.01(3H,t,J=7.3Hz),1.48(9H,s),4.16(2H,q,J=7.5Hz),4.22-4.28(2H,m),4.43-4.56(2H,m),6.09(1H,s),7.47(2H,s)。
[步骤3]5-[1-(叔丁氧基羰基)-3-甲氧基氮杂环丁烷-3-基]-3-(2,4,6-三氯苯基)-1,2-唑-4-甲酸乙基酯
使用在上述步骤2获得的化合物(876mg),通过与参考实施例X-67的步骤3相同的方法,得到标题化合物(732mg)。
1H-NMR(CDCl3)δ:1.10(3H,t,J=7.0Hz),1.47(9H,s),3.20(3H,s),4.16(2H,q,J=7.3Hz),4.31(2H,d,J=10.3Hz),4.52(2H,d,J=10.3Hz),7.46(2H,s)。
[步骤4]5-(3-甲氧基氮杂环丁烷-3-基)-3-(2,4,6-三氯苯基)-1,2-唑-4-甲酸乙基酯盐酸盐
使用上述步骤3获得的化合物(732mg),通过与参考实施例X-62的步骤1相同的方法,得到标题化合物,直接用于下一反应。
[步骤5]5-[3-甲氧基-1-(2-硝基苯基)磺酰基氮杂环丁烷-3-基]-3-(2,4,6-三氯苯基)-1,2-唑-4-甲酸乙基酯
使用上述步骤4获得的化合物,通过与参考实施例X-62的步骤2相同的方法,得到标题化合物(831mg)。
1H-NMR(CDCl3)δ:1.11(3H,t,J=7.3Hz),3.17(3H,s),4.17(2H,q,J=7.3Hz),4.54(2H,d,J=9.7Hz),4.76(2H,d,J=9.7Hz),7.46(2H,s),7.71-7.76(3H,m),8.07-8.12(1H,m)。
[步骤6]5-[3-甲氧基-1-(2-硝基苯基)磺酰基氮杂环丁烷-3-基]-3-(2,4,6-三氯苯基)-1,2-唑-4-甲酸
使用上述步骤5获得的化合物(831mg),通过与参考实施例X-1步骤4相同的方法,得到标题化合物(674mg)。
1H-NMR(CDCl3)δ:3.20(3H,s),4.53(2H,d,J=9.7Hz),4.75(2H,d,J=9.7Hz),7.47(2H,s),7.72-7.78(3H,m),8.07-8.12(1H,m)。
[参考实施例X-74]3-(2,6-二氯-4-环丙基苯基)-5-(2-氟丙烷-2-基)-1,2-唑-4-甲酸
式51
[步骤1]3-(2,6-二氯-4-环丙基苯基)-5-(2-氟丙烷-2-基)-1,2-唑-4-甲酸乙基酯
向参考实施例X-49获得的化合物(1.69g)、三环己基膦(200mg)、环丙基硼酸(368mg)和磷酸三钾(2.27g)的甲苯(17.8ml)和水(3.6ml)悬浮液中加入乙酸钯(80mg),将混合物在100℃搅拌3小时。冷却后,向反应液中加入乙酸乙酯和水,分离两层。将有机层用水和饱和盐水依次洗涤,经无水硫酸钠干燥。减压蒸除溶剂,将获得的残留物经硅胶柱色谱(正己烷/乙酸乙酯)进行纯化,得到标题化合物(1.09g)。
1H-NMR(CDCl3)δ:0.74-0.78(2H,m),1.01(3H,t,J=7.3Hz),1.05-1.10(2H,m),1.86-1.99(7H,m),4.11(2H,q,J=7.3Hz),7.09(2H,s)。
MS(m/z):386(M+H)+。
[步骤2]3-(2,6-二氯-4-环丙基苯基)-5-(2-氟丙烷-2-基)-1,2-唑-4-甲酸
使用在上述步骤1获得的化合物(1.09g),通过与参考实施例X-1步骤4相同的方法,得到标题化合物(655mg)。
1H-NMR(CDCl3)δ:0.78(2H,ddd,J=5.7,4.2,2.1Hz),1.05-1.11(2H,m),1.87-1.99(7H,m),7.09(2H,s)。
[参考实施例X-75]3-(叔丁基)-5-(2,4,6-三氯苯基)-1,2-唑-4-甲酸
式52
[步骤1]1,3,5-三氯-2-乙炔基苯
于2,4,6-三氯苯甲醛(1.68g)的甲醇(80ml)溶液中加入碳酸钾(2.22g)和(1-二偶氮-2-氧代丙基)膦酸二甲基酯(1.44ml),于室温下将混合物搅拌两天。在冰冷却下,向反应液加入饱和碳酸氢钠水溶液,将混合物减压浓缩。向获得的残留物中加入乙酸乙酯,用水和饱和盐水依次洗涤,经无水硫酸钠干燥。减压蒸除溶剂,将获得的残留物经硅胶柱色谱(正己烷/乙酸乙酯)进行纯化,得到标题化合物(1.35g)。
1H-NMR(CDCl3)δ:3.71(1H,s),7.37(2H,s)。
[步骤2]3-(2,4,6-三氯苯基)丙-2-炔酸乙基酯
在-78℃下,向上述步骤1获得的化合物(1.35g)的四氢呋喃(14ml)溶液中加入n-丁基锂(1.58M的正己烷溶液,4.6ml),将混合物在与上述相同的温度下搅拌30分钟。向反应液加入氯甲酸乙酯(0.82ml),将混合物在与上述相同的温度下搅拌1.5小时,倾至乙酸乙酯和饱和的氯化铵水溶液中并剧烈搅拌。分离两层,将有机层用水和饱和盐水依次洗涤,经无水硫酸钠干燥。减压蒸除溶剂,将获得的残留物经硅胶柱色谱(正己烷/乙酸乙酯)进行纯化,得到标题化合物(732mg)。
1H-NMR(CDCl3)δ:1.37(3H,t,J=7.3Hz),4.33(2H,q,J=7.1Hz),7.39(2H,s)。
[步骤3](1E)-2,2-二甲基丙醛肟
在冰冷却下,向2,2-二甲基丙醛(2.0ml)的乙醇(11ml)溶液中加入羟基胺盐酸盐(1.4g)和1N氢氧化钠水溶液(45ml),于室温下将混合物搅拌2小时。在冰冷却下,向反应液加入浓盐酸使其呈酸性,用乙醚进行萃取。将萃取液用饱和盐水洗涤,经无水硫酸钠干燥,经减压蒸除溶剂后得到标题化合物(790mg)。
1H-NMR(CDCl3)δ:1.11(9H,s),7.35(1H,s),7.77(1H,brs)。
[步骤4](1Z)-N-羟基-2,2-二甲基丙亚氨酰氯(propanimidoyl chloride)
向上述步骤3获得的化合物(303mg)的N,N-二甲基甲酰胺(6ml)溶液中加入N-氯琥珀酸酰亚胺(401mg),于室温下将混合物搅拌过夜。将反应液用乙醚稀释,用水和饱和盐水依次洗涤,经无水硫酸钠干燥。经减压蒸除溶剂后得到标题化合物(473mg)。
1H-NMR(CDCl3)δ:1.26(9H,s),7.52(1H,s)。
[步骤5]3-(叔丁基)-5-(2,4,6-三氯苯基)-1,2-唑-4-甲酸乙基酯
向上述步骤2获得的化合物(278mg)与上述步骤4获得的化合物(407mg)的N,N-二甲基甲酰胺(5ml)溶液中缓慢加入三乙基胺(0.83ml),于室温下将混合物搅拌过夜后,在50℃搅拌6小时。将反应液用乙酸乙酯稀释,用水和饱和盐水依次洗涤,经无水硫酸钠干燥。减压蒸除溶剂,将获得的残留物经硅胶柱色谱(正己烷/乙酸乙酯)进行纯化,得到标题化合物(406mg)。
1H-NMR(CDCl3)δ:1.00(3H,t,J=7.0Hz),1.52(9H,s),4.12(3H,q,J=7.1Hz),7.45(2H,s)。
[步骤6]3-(叔丁基)-5-(2,4,6-三氯苯基)-1,2-唑-4-甲酸
使用上述步骤5获得的化合物(406mg),通过与参考实施例X-1步骤4相同的方法,得到标题化合物(211mg)。
1H-NMR(CDCl3)δ:1.52(9H,s),7.45(2H,s)。
通过与参考实施例X-75相同的方法,得到下述化合物。
[表26]
[参考实施例E-1](E)-3-(1H-吲哚-4-基)丙-2-烯酸叔丁基酯
式53
[步骤1]
向1H-吲哚-4-甲醛(30g)的N,N-二甲基甲酰胺(300ml)溶液中加入二乙基膦酰基乙酸叔丁基酯(53.4ml)和碳酸钾(60g),将混合物在80℃下搅拌9.5小时。冷却后,向反应液加入水,用乙酸乙酯进行萃取。将萃取液用水和饱和盐水依次洗涤,经无水硫酸钠干燥。减压蒸除溶剂,将获得的残留物经硅胶柱色谱(正己烷/乙酸乙酯)进行纯化。减压浓缩洗脱的组分,向获得的残留物中加入正己烷,于室温下将混合物进行搅拌后,过滤收集悬浮液,得到标题化合物(41.3g)。
1H-NMR(CDCl3)δ:1.56(9H,s),6.55(1H,d,J=16.3Hz),6.84(1H,brs),7.20(1H,t,J=7.9Hz),7.32(1H,t,J=3.0Hz),7.37(1H,d,J=7.3Hz),7.43(1H,d,J=7.9Hz),8.01(1H,d,J=15.7Hz),8.32(1H,brs)。
通过与参考实施例E-1相同的方法,得到下述化合物。
[表27]
[参考实施例E-3](E)-3-(3-氟-1H-吲哚-4-基)丙-2-烯酸叔丁基酯
式54
[步骤1]
向参考实施例E-1的步骤1获得的化合物(1.60g)的丙酮(13ml)和乙腈(20ml)混合液中加入1-氯甲基-4-氟-1,4-二氮杂双环[2.2.2]辛烷(diazoniabicyclo[2.2.2.]octane)双(四氟硼酸盐)(2.70g),于室温下将混合物搅拌1小时。将反应液减压浓缩,将获得的残留物用乙酸乙酯稀释,用饱和碳酸氢钠水溶液、水(50ml)和饱和盐水(50ml)依次洗涤,经无水硫酸钠干燥。减压蒸除溶剂,将获得的残留物经硅胶柱色谱(正己烷/乙酸乙酯)进行纯化,得到标题化合物(74mg)。
1H-NMR(CDCl3)δ:1.56(9H,s),6.51(1H,dd,J=15.7,1.8Hz),7.07(1H,dd,J=2.7,1.4Hz),7.21(1H,dd,J=7.9,3.9Hz),7.32(1H,dd,J=8.2,2.7Hz),7.44(1H,d,J=7.3Hz),7.70(1H,brs),8.26(1H,d,J=15.7Hz)。
MS(m/z):260(M-H)-。
[参考实施例E-4](E)-3-(3-氯-1H-吲哚-4-基)丙-2-烯酸叔丁基酯
式55
[步骤1]
向参考实施例E-1的步骤1获得的化合物(1.20g)的N,N-二甲基甲酰胺(20ml)溶液中加入N-氯琥珀酸酰亚胺(659mg),于室温下将混合物搅拌2小时。将反应液用乙酸乙酯稀释,用水和饱和盐水依次洗涤,经无水硫酸镁干燥。减压蒸除溶剂,将获得的残留物经硅胶柱色谱(正己烷/乙酸乙酯)进行纯化,得到标题化合物(1.25g)。
1H-NMR(CDCl3)δ:1.56(9H,s),6.41(1H,d,J=15.7Hz),7.20-7.25(3H,m),7.38(1H,d,J=7.9Hz),8.21(1H,brs),8.90(1H,d,J=15.7Hz)。
通过与参考实施例E-4相同的方法,得到下述化合物。
[表28]
[参考实施例E-6](E)-3-(3-甲酰基-1H-吲哚-4-基)丙-2-烯酸叔丁基酯
式56
[步骤1]
在氮环境中、于冰冷却下,向N,N-二甲基甲酰胺(0.883g)的1,2-二氯乙烷(65ml)溶液中加入草酰氯(1.3ml)的1,2-二氯乙烷(7ml)溶液,将混合物在与上述相同的温度下搅拌20分钟。向反应液加入参考实施例E-1的步骤1获得的化合物(2.45g),于室温下将混合物搅拌3.5小时。在冰冷却下向反应液中加入10%碳酸钠水溶液(50ml),将混合物在与上述相同的温度下搅拌2小时。分离两层,将水层用二氯甲烷进行萃取。合并有机层,用饱和盐水洗涤,经无水硫酸钠干燥。减压蒸除溶剂,将获得的残留物经硅胶柱色谱(正己烷/乙酸乙酯)进行纯化,得到标题化合物(1.99g)。
1H-NMR(CDCl3)δ:1.59(9H,s),6.41(1H,d,J=15.1Hz),7.32(1H,dd,J=7.9,3.9Hz),7.47(1H,d,J=8.5Hz),7.61(1H,d,J=7.9Hz),8.01(1H,d,J=3.0Hz),9.21(2H,d,J=16.3Hz),10.03(1H,s)。
MS(m/z):270(M-H)-。
[参考实施例E-7]2-(1H-吲哚-4-基)环丙烷羧酸叔丁基酯
式57
[步骤1]
将碘化三甲基氧化锍溶于二甲基亚砜(2ml),向该溶液中加入叔丁醇钾(138mg),于室温下将混合物搅拌1小时。向反应溶液加入参考实施例E-1的步骤1获得的化合物(150mg),于室温下将混合物搅拌17小时。向反应液加入饱和盐水后,用乙酸乙酯萃取,将萃取液用饱和盐水洗涤,经无水硫酸钠干燥。减压蒸除溶剂,将获得的残留物经硅胶柱色谱(正己烷/乙酸乙酯)进行纯化,得到标题化合物(66.9mg)。
1H-NMR(CDCl3)δ:1.37-1.43(1H,m),1.50(9H,s),1.56-1.62(1H,m),1.93-1.99(1H,m),2.75-2.81(1H,m),6.67-6.70(1H,m),6.73-6.77(1H,m),7.09-7.14(1H,m),7.22-7.25(1H,m),7.25-7.29(1H,m),8.23(1H,br)。
MS(ESI):258(M+H)+。
[参考实施例E-8](E)-3-(3-甲基-1H-吲哚-4-基)丙-2-烯酸叔丁基酯
式58
[步骤1]
向4-溴-3-甲基-1H-吲哚(15g)的N,N-二甲基甲酰胺(500ml)溶液中加入N,N-二(丙烷-2-基)乙基胺(18.7ml)、三(2-甲基苯基)膦(2.2g)、乙酸钯(0.8g)和丙烯酸叔丁基酯(15.7ml),在氮环境,140℃将混合物搅拌8小时。冷却后,向反应液中加入水,用乙酸乙酯进行萃取。将萃取液用水和饱和盐水依次洗涤,经无水硫酸钠干燥。减压蒸除溶剂,将获得的残留物经硅胶柱色谱(正己烷/乙酸乙酯)进行纯化。减压浓缩洗脱的组分,向获得的残留物中加入正己烷,于室温下搅拌混合物,将悬浮液过滤,收集得到标题化合物(15.6g)。
1H-NMR(CDCl3)δ:1.56(9H,s),2.56(3H,s),6.39(1H,d,J=15.7Hz),7.03(1H,s),7.15(1H,t,J=7.3Hz),7.35(1H,d,J=8.5Hz),7.41(1H,d,J=7.3Hz),7.99(1H,brs),8.49(1H,d,J=15.7Hz)。
通过与参考实施例E-8相同的方法,得到下述化合物。
[表29]
[参考实施例E-13]3-(3-甲基-1H-吲哚-4-基)丙酸叔丁基酯
式59
[步骤1]
在氮环境下,向参考实施例E-8的步骤1获得的化合物(364mg)的乙酸乙酯(10ml)溶液中加入10%钯炭催化剂(湿的,152mg),将混合物在氢气环境中、于室温下搅拌2.5小时。将反应液通过硅藻土过滤,将滤液减压浓缩。将获得的残留物经硅胶柱色谱(正己烷/乙酸乙酯)进行纯化,得到标题化合物(306mg)。
1H-NMR(CDCl3)δ:1.45(9H,s),2.51(3H,d,J=1.2Hz),2.58-2.64(2H,m),3.31-3.37(2H,m),6.87(1H,d,J=7.3Hz),6.94(1H,q,J=1.2Hz),7.07(1H,t,J=7.6Hz),7.20(1H,d,J=7.9Hz),7.88(1H,brs)。
[参考实施例E-14](E)-3-(7-氟-3-甲基-1H-吲哚-4-基)丙-2-烯酸叔丁基酯
式60
[步骤1]4-溴-7-氟-1H-吲哚-3-甲醛
使用4-溴-7-氟-1H-吲哚(1.10g),通过与参考实施例E-6的步骤1相同的方法,得到标题化合物(502mg)。
1H-NMR(CDCl3)δ:6.91(1H,dd,J=10.0,8.2Hz),7.40(1H,dd,J=8.5,4.2Hz),8.11(1H,d,J=3.0Hz),9.10(1H,brs),10.92(1H,s)。
MS(m/z):242(M+H)+。
[步骤2]4-溴-7-氟-3-甲基-1H-吲哚
在氮气环境中、在冰冷却下,向上述步骤1获得的化合物(500mg)的甲苯(10ml)和四氢呋喃(2ml)混合液中加入氢化双(2-甲氧基乙氧基)铝钠(65%甲苯溶液,1.89ml),将混合物加热至回流1小时。在冰冷却下,向反应液中加入饱和氯化铵水溶液,用乙酸乙酯进行萃取。将萃取液用饱和盐水洗涤,经无水硫酸钠干燥。减压蒸除溶剂,将获得的残留物经硅胶柱色谱(正己烷/乙酸乙酯)进行纯化,得到标题化合物(357mg)。
1H-NMR(CDCl3)δ:2.54(3H,s),6.73(1H,dd,J=10.3,8.5Hz),7.00-7.03(1H,brm),7.12(1H,dd,J=8.5,4.2Hz),8.12(1H,brs)。
MS(m/z):228(M+H)+。
[步骤3](E)-3-(7-氟-3-甲基-1H-吲哚-4-基)丙-2-烯酸叔丁基酯
使用在上述步骤2获得的化合物(354mg),通过与参考实施例E-8的步骤1相同的方法,得到标题化合物(366mg)。
1H-NMR(CDCl3)δ:1.55(9H,s),2.54(3H,s),6.33(1H,d,J=15.7Hz),6.87(1H,dd,J=10.6,8.2Hz),7.05-7.05(1H,brm),7.33(1H,dd,J=8.5,4.8Hz),8.14(1H,brs),8.40(1H,d,J=15.7Hz)。
[参考实施例E-15](E)-3-(3-乙酰基-1H-吲哚-4-基)丙-2-烯酸叔丁基酯
式61
[步骤1]1-(4-溴-1H-吲哚-3-基)乙酮
在冰冷却下,向4-溴-1H-吲哚(1.96g)的二氯甲烷(20ml)溶液中加入乙酰氯(1.18g)和四氯化锡(1.0M的二氯甲烷溶液,15ml),于室温下将混合物搅拌1小时。向反应液加入饱和碳酸氢钠水溶液和硅藻土,于室温下搅拌混合物30分钟,然后过滤。分离两层,将有机层用饱和盐水洗涤,经无水硫酸镁干燥。减压蒸除溶剂,将获得的固体用二氯甲烷/正己烷洗涤,得到标题化合物(2.02g)。
1H-NMR(CDCl3)δ:2.62(3H,s),7.11-7.14(1H,m),7.36-7.38(1H,m),7.48-7.51(1H,m),7.79-7.81(1H,m),8.63(1H,brs)。
[步骤2](E)-3-(3-乙酰基-1H-吲哚-4-基)丙-2-烯酸叔丁基酯
使用在上述步骤1获得的化合物(2.02g),通过与参考实施例E-8的步骤1相同的方法,得到标题化合物(2.02g)。
1H-NMR(CDCl3)δ:1.57(9H,s),2.58(3H,s),6.31(1H,d,J=15.7Hz),7.29(1H,d,J=7.9Hz),7.42(1H,d,J=8.5Hz),7.55(1H,d,J=7.9Hz),7.98-7.99(1H,m),8.83(1H,s),9.24(1H,d,J=15.7Hz)。
[参考实施例E-16](E)-3-(3-乙基-1H-吲哚-4-基)丙-2-烯酸叔丁基酯
式62
[步骤1]4-溴-1-[(4-甲基苯基)磺酰基]-1H-吲哚
在冰冷却下,向4-溴-1H-吲哚(6.18g)的甲苯(50ml)溶液中加入50%氢氧化钠水溶液(24.6g)和对甲苯磺酰氯(6.4g),于室温下将混合物搅拌过夜。向反应液中加入水,分离两层,将有机层用饱和盐水洗涤,经无水硫酸镁干燥。减压蒸除溶剂,将获得的固体用二氯甲烷/正己烷洗涤,得到标题化合物(9.33g)。
1H-NMR(CDCl3)δ:2.35(3H,s),6.72(1H,d,J=3.6Hz),7.16-7.18(1H,m),7.24(2H,d,J=7.9Hz),7.39(1H,d,J=7.9Hz),7.62(1H,d,J=3.6Hz),7.76(2H,d,J=7.9Hz),7.94(1H,d,J=8.5Hz)。
[步骤2]1-[4-溴-1-[(4-甲基苯基)磺酰基]-1H-吲哚-3-基]乙酮
在冰冷却下,向二氯甲烷(60ml)中加入氯化铝(10.7g)和乙酸酐(5.44g)。确认氯化铝已溶解后,加入上述步骤1获得的化合物(9.33g),于室温下将混合物搅拌1小时。将反应液用二氯甲烷稀释,用水、饱和碳酸氢钠水溶液和饱和盐水依次洗涤,然后经无水硫酸镁干燥。减压蒸除溶剂,将获得的固体用二氯甲烷/正己烷洗涤,得到标题化合物(9.15g)。
1H-NMR(CDCl3)δ:2.38(3H,s),2.64(3H,s),7.20-7.22(1H,m),7.28(2H,d,J=8.5Hz),7.52(1H,d,J=7.9Hz),7.79(2H,d,J=8.5Hz),7.95(1H,d,J=8.5Hz),8.04(1H,s)。
[步骤3]4-溴-3-乙基-1-[(4-甲基苯基)磺酰基]-1H-吲哚
在冰冷却下,向三氟乙酸(20ml)和二氯甲烷(15ml)的混合液中加入上述步骤2获得的化合物(8.01g)和硼氢化钠(7.72g),于室温下将混合物搅拌4小时。将反应液用二氯甲烷稀释,用水和饱和盐水依次洗涤,经无水硫酸镁干燥。减压蒸除溶剂,将获得的残留物经硅胶柱色谱(正己烷/乙酸乙酯/二氯甲烷)进行纯化,得到标题化合物(6.61g)。
1H-NMR(CDCl3)δ:1.31(3H,t,J=7.3Hz),2.35(3H,s),2.98(2H,q,J=7.3Hz),7.09-7.11(1H,m),7.22(2H,d,J=7.9Hz),7.36-7.37(2H,m),7.73(2H,d,J=7.9Hz),7.94(1H,d,J=9.1Hz)。
[步骤4]4-溴-3-乙基-1H-吲哚
于室温下将上述步骤3获得的化合物(1.10g)、镁(0.29g)、氯化铵(83.8mg)和甲醇(20ml)的混合物搅拌2小时。向反应液加入饱和氯化铵水溶液,用乙酸乙酯进行萃取。将萃取液用水和饱和盐水依次洗涤,经无水硫酸镁干燥。减压蒸除溶剂,将获得的残留物经硅胶柱色谱(正己烷/乙酸乙酯)进行纯化,得到标题化合物(0.51g)。
1H-NMR(CDCl3)δ:1.33(3H,t,J=7.5Hz),3.07(2H,q,J=7.5Hz),6.97-7.02(2H,m),7.26-7.29(2H,m),8.01(1H,brs)。
[步骤5](E)-3-(3-乙基-1H-吲哚-4-基)丙-2-烯酸叔丁基酯
使用上述步骤4获得的化合物(1.23g),通过与参考实施例E-8的步骤1相同的方法,得到标题化合物(1.21g)。
1H-NMR(CDCl3)δ:1.37(3H,t,J=7.5Hz),1.56(9H,s),3.00(2H,q,J=7.5Hz),6.38(1H,d,J=15.7Hz),7.05-7.06(1H,m),7.15-7.17(1H,m),7.35-7.40(2H,m),8.02(1H,brs),8.44(1H,d,J=15.7Hz)。
[参考实施例E-17](2E)-(3,4-二氢0苯并[cd]吲哚-5(1H)-亚基乙酸叔丁基酯
式63
[步骤1](2E)-[1-(2,2-二甲基丙酰基)-3,4-二氢苯并[cd]吲哚-5(1H)-亚基乙酸叔丁基酯
在冰冷却下,向二乙基膦酰乙酸叔丁基酯(1.12ml)的四氢呋喃(5ml)溶液中加入氢化钠(55%的油分散液,190mg),将混合物搅拌30分钟。向反应液加入1-(2,2-二甲基丙酰基)-3,4-二氢苯并[cd]吲哚-5(1H)-酮[Tetrahedron Lett.,39,8729-8732(1998)](1.01g)的四氢呋喃(10ml)溶液,于室温下将混合物搅拌过夜。将反应液用乙酸乙酯稀释,用水和饱和盐水依次洗涤,经无水硫酸钠干燥。减压蒸除溶剂,将获得的残留物经硅胶柱色谱(正己烷/乙酸乙酯)进行纯化,得到标题化合物(490mg)。
1H-NMR(CDCl3)δ:1.52(9H,s),1.55(9H,s),2.95(2H,t,J=6.7Hz),3.43(2H,t,J=6.7Hz),6.44(1H,s),7.33(1H,t,J=7.9Hz),7.43-7.48(2H,m),8.33(1H,d,J=7.9Hz)。
[步骤2](2E)-(3,4-二氢-1H-苯并[cd]吲哚-5(1H)-亚基乙酸叔丁基酯
于室温下向上述步骤1获得的化合物(490mg)的四氢呋喃(1ml)和甲醇(3ml)混合液中加入1N氢氧化钠水溶液(3ml),于室温下将混合物搅拌1小时。将反应液用乙酸乙酯稀释,用1N盐酸、水和饱和盐水依次洗涤,经无水硫酸钠干燥。减压蒸除溶剂,将获得的残留物经硅胶柱色谱(正己烷/乙酸乙酯)进行纯化,得到标题化合物(298mg)。
1H-NMR(CDCl3)δ:1.55(9H,s),2.98-3.03(2H,m),3.43-3.49(2H,m),6.45(1H,t,J=1.5Hz),6.94-6.95(1H,m),7.18(1H,t,J=7.9Hz),7.28-7.33(2H,m),7.93(1H,brs)。
[参考实施例E-18]1,3,4,5-四氢苯并[cd]吲哚-5-基乙酸叔丁基酯
式64
[步骤1]
使用参考实施例E-17的步骤2获得的化合物(149mg),通过与参考实施例E-13的步骤1相同的方法,得到标题化合物(90mg)。
1H-NMR(CDCl3)δ:1.49(9H,s),1.86-1.96(1H,m),2.08-2.21(1H,m),2.44(1H,dd,J=14.8,8.8Hz),2.79(1H,dd,J=14.5,6.0Hz),2.82-2.97(2H,m),3.47-3.58(1H,m),6.85-6.91(2H,m),7.12(1H,t,J=7.6Hz),7.18(1H,d,J=7.9Hz),7.86(1H,brs)。
[参考实施例E-19](2E)-(6-氟-3,4-二氢-1H-苯并[cd]吲哚-5(1H)-亚基乙酸叔丁基酯
式65
[步骤1]3-(5-氟-1H-吲哚-3-基)丙烷酸
将5-氟-1H-吲哚(2.03g)、乙酸(15ml)和丙烯酸(2.27ml)的混合物在90℃搅拌3天。将反应液减压浓缩,向获得的残留物中加入3N氢氧化钠水溶液,搅拌混合物,过滤不溶物并除去。将滤液用乙醚洗涤,在冰冷却下向获得的水层中加入浓盐酸使其呈酸性,用二氯甲烷进行萃取。将萃取液经无水硫酸镁干燥。经减压蒸除溶剂后得到标题化合物(2.37g)。
1H-NMR(CDCl3)δ:2.76(2H,t,J=7.6Hz),3.07(2H,t,J=7.6Hz),6.90-6.99(1H,m),7.07(1H,d,J=2.4Hz),7.22-7.28(2H,m),7.98(1H,brs)。
[步骤2]3-[1-(2,2-二甲基丙酰基)-5-氟-1H-吲哚-3-基]丙烷酸
在氮气流中-78℃下,向上述步骤1获得的化合物(2.37g)的四氢呋喃(57ml)溶液中加入n-丁基锂(1.6M的正己烷溶液,14.6ml),将混合物在与上述相同的温度下搅拌5分钟。向反应液加入新戊酰氯(1.51ml),于室温下将混合物搅拌3小时。在冰冷却下,向反应液中加入饱和氯化铵水溶液,用乙酸乙酯进行萃取。将萃取液用饱和盐水洗涤,经无水硫酸镁干燥。减压蒸除溶剂后,得到粗品标题化合物(3.21g),无须纯化直接使用于下一反应。
[步骤3]1-(2,2-二甲基丙酰基)-6-氟-3,4-二氢苯并[cd]吲哚-5(1H)-酮
在氮气流中、冰冷却下,向上述步骤2获得的化合物(2.85g)的二氯甲烷(24.5ml)溶液中加入亚硫酰氯(2.86ml),于室温下将混合物搅拌20分钟。将反应液减压浓缩,将获得的残留物溶于二氯甲烷(24.5ml),得到酰氯溶液。
在氮气流中、冰冷却下,向氯化铝(2.87g)的二氯甲烷(24.5ml)溶液中滴加氯代乙酰氯(2.57ml)。随后向加入制备的酰氯溶液,于室温下将混合物搅拌20分,然后加热至回流2.5小时。将反应液加至冰中,用二氯甲烷萃取,将萃取液经无水硫酸镁干燥。减压蒸除溶剂,将获得的残留物经硅胶柱色谱(正己烷/乙酸乙酯)进行纯化,得到标题化合物(662mg)。
1H-NMR(CDCl3)δ:1.53(9H,s),2.89(2H,t,J=7.3Hz),3.20(2H,q,J=4.8Hz),7.10(1H,dd,J=10.9,9.1Hz),7.63(1H,s),8.50(1H,dd,J=8.8,3.9Hz)。
[步骤4](2E)-[1-(2,2-二甲基丙酰基)-6-氟-3,4-二氢-1H-苯并[cd]吲哚-5(1H)-亚基乙酸叔丁基酯
使用上述步骤3获得的化合物(662mg),通过与参考实施例E-17的步骤1相同的方法,得到标题化合物(192mg)。
1H-NMR(CDCl3)δ:1.51(9H,s),1.54(9H,s),2.91(2H,t,J=6.3Hz),3.39(2H,t,J=6.7Hz),6.69(1H,s),7.06(1H,dd,J=11.8,8.8Hz),7.48(1H,s),8.25(1H,dd,J=9.1,3.6Hz)。
[步骤5](2E)-(6-氟-3,4-二氢-1H-苯并[cd]吲哚-5(1H)-亚基乙酸叔丁基酯
使用上述步骤4获得的化合物(192mg),通过与参考实施例E-17的步骤2相同的方法,得到标题化合物(146mg)。
1H-NMR(CDCl3)δ:1.55(9H,s),2.95(2H,t,J=6.7Hz),3.42(2H,t,J=6.3Hz),6.70(1H,s),6.89-6.99(2H,m),7.19(1H,dd,J=8.5,3.0Hz),7.91(1H,brs)。
[参考实施例E-20]6,7,8,9-四氢-3H-苯并[e]吲哚-8-甲酸叔丁基酯
式66
[步骤1]5-[(1E)-4-甲氧基-4-氧代丁-1-烯-1-基]-1H-吲哚-4-甲酸甲基酯
向5-溴-1H-吲哚-4-甲酸甲基酯(WO2004/063198)(1.21g)的N,N-二甲基甲酰胺(9.5ml)溶液中,加入N,N-二(丙烷-2-基)乙基胺(2.49ml)、三(2-甲基苯基)膦(580mg)、乙酸钯(214mg)和丁-3-烯酸甲基酯(1.07ml),使用微波装置,将混合物在氮环境下于140℃搅拌1小时。将混合物用乙酸乙酯稀释,用水和饱和盐水依次洗涤,经无水硫酸钠干燥。减压蒸除溶剂,将获得的残留物经硅胶柱色谱(正己烷/乙酸乙酯)进行纯化,得到标题化合物(990mg)。
1H-NMR(CDCl3)δ:3.32(2H,dd,J=7.3,1.8Hz),3.73(3H,s),4.00(3H,s),6.20(1H,dt,J=15.7,7.3Hz),6.83(1H,t,J=2.7Hz),7.02-7.53(4H,m),8.29(1H,brs)。
[步骤2]5-(4-甲氧基-4-氧代丁基)-1H-吲哚-4-甲酸甲基酯
使用在上述步骤1获得的化合物(990mg),通过与参考实施例E-13的步骤1相同的方法,得到标题化合物(850mg)。
1H-NMR(CDCl3)δ:1.99(2H,tt,J=7.9,7.9Hz),2.38(2H,t,J=7.6Hz),3.01(2H,t,J=7.6Hz),3.66(3H,s),3.99(3H,s),6.81-6.84(1H,m),7.08(1H,d,J=8.5Hz),7.28(1H,t,J=2.4Hz),7.44(
1H,d,J=8.5Hz),8.23(1H,brs)。
[步骤3]5-(4-甲氧基-4-氧代丁基)吲哚-1,4-二羧酸1-叔丁基4-甲基酯
在冰冷却下,向上述步骤2获得的化合物(850mg)的二氯甲烷(15ml)溶液中加入三乙基胺(0.86ml)、4-二甲基氨基吡啶(38mg)和二碳酸二叔丁基酯(809mg),于室温下将混合物搅拌3天。将反应液减压浓缩,将获得的残留物经硅胶柱色谱(正己烷/乙酸乙酯)进行纯化,得到标题化合物(1.00g)。
1H-NMR(CDCl3)δ:1.67(9H,s),1.93-2.02(2H,m),2.37(2H,t,J=7.6Hz),2.98(2H,t,J=7.6Hz),3.67(3H,s),3.98(3H,s),6.84(1H,d,J=3.6Hz),7.19(1H,d,J=9.1Hz),7.62(1H,d,J=3.6Hz),8.20(1H,d,J=8.5Hz)。
[步骤4]9-氧代-6,7,8,9-四氢-3H-苯并[e]吲哚-3,8-二羧酸3-叔丁基8-甲基酯
于-78℃,向二(丙烷-2-基)氨化锂(1.09M的四氢呋喃溶液,7.33ml)溶液中滴加上述步骤3获得的化合物(1.00g)的四氢呋喃(5ml)溶液,将混合物搅拌2小时。将反应液倾至乙酸乙酯与1N盐酸的混合液中,剧烈搅拌,分离两层。将有机层用饱和盐水洗涤,经无水硫酸钠干燥。减压蒸除溶剂,将获得的残留物经硅胶柱色谱(正己烷/乙酸乙酯)进行纯化,得到标题化合物(809mg)。
[步骤5]9-氧代-6,7,8,9-四氢-3H-苯并[e]吲哚-8-甲酸甲基酯
在冰冷却下,向上述步骤4获得的化合物(809mg)的二氯甲烷(7.9ml)溶液中加入三氟乙酸(7.9ml),于室温下将混合物搅拌1小时。将反应液减压浓缩,得到标题化合物(695mg)。
1H-NMR(CDCl3)δ:2.38-2.48(1H,m),2.52-2.64(1H,m),3.06-3.24(2H,m),3.74(1H,dd,J=10.3,4.8Hz),3.82(3H,s),7.23-7.28(1H,m),7.65(1H,d,J=3.6Hz),7.75(1H,d,J=3.6Hz),8.36(1H,d,J=8.5Hz)。
[步骤6]6,7,8,9-四氢-3H-苯并[e]吲哚-8-甲酸甲基酯
通过向上述步骤5获得的化合物(547mg)中加入乙酸乙酯(5ml)、甲醇(10ml)、二氯甲烷(3ml)和乙酸(0.13ml)使其溶解,然后在氮环境下,加入5%钯炭催化剂(湿的,547mg)。将混合物在氢环境下、于50℃搅拌7小时,充入氮,用硅藻土过滤。将滤液减压浓缩,将获得的残留物经硅胶柱色谱(正己烷/乙酸乙酯)进行纯化,得到标题化合物(111mg)。
1H-NMR(CDCl3)δ:1.86-1.98(1H,m),2.24-2.33(1H,m),2.85(1H,tdd,J=11.2,5.5,2.9Hz),2.92-2.99(2H,m),3.15(1H,dd,J=16.6,11.2Hz),3.31(1H,dd,J=16.9,5.4Hz),3.76(3H,s),6.53(1H,t,J=2.4Hz),6.94(1H,d,J=8.5Hz),7.17-7.22(2H,m),8.13(1H,brs)。
[步骤7]6,7,8,9-四氢-3H-苯并[e]吲哚-8-甲酸
于室温下向上述步骤6获得的化合物(111mg)的四氢呋喃(1ml)和甲醇(1ml)混合液中加入1N氢氧化钠水溶液(1ml),将混合物在50℃搅拌1小时。将反应液减压浓缩,向获得的残留物中加入乙酸乙酯和1N盐酸,分离两层。将有机层用饱和盐水洗涤,经无水硫酸钠干燥。经减压蒸除溶剂后得到标题化合物(104mg)。
1H-NMR(CDCl3)δ:1.90-2.02(1H,m),2.29-2.38(1H,m),2.91(1H,tdd,J=10.9,5.5,2.9Hz),2.95-3.02(2H,m),3.18(1H,dd,J=16.9,10.3Hz),3.34(1H,dd,J=16.9,5.4Hz),6.53(1H,t,J=2.4Hz),6.95(1H,d,J=8.5Hz),7.18-7.23(2H,m),8.14(1H,brs)。
[步骤8]6,7,8,9-四氢-3H-苯并[e]吲哚-8-甲酸叔丁基酯
于70℃向上述步骤7获得的化合物(104mg)的甲苯(5ml)悬浮液中加入N,N-二甲基甲酰胺二-叔丁基缩醛(1.2ml),将混合物在90℃搅拌45分钟。冷却后,将反应液用乙酸乙酯稀释,用水和饱和盐水依次洗涤、然后经无水硫酸钠干燥。减压蒸除溶剂,将获得的残留物经硅胶柱色谱(正己烷/乙酸乙酯)进行纯化,得到标题化合物(82mg)。
1H-NMR(CDCl3)δ:1.50(9H,s),1.80-1.93(1H,m),2.20-2.28(1H,m),2.72(1H,tdd,J=11.2,5.4,2.8Hz),2.90-2.97(2H,m),3.10(1H,dd,J=16.6,10.6Hz),3.24(1H,dd,J=16.9,5.4Hz),6.53(1H,t,J=2.4Hz),6.93(1H,d,J=8.5Hz),7.16-7.21(2H,m),8.12(1H,brs)。
[实施例1](2E)-3-(1-{[5-(3-甲基氧杂环丁烷-3-基)-3-(2,4,6-三氯苯基)-1,2-唑-4-基]羰基}-1H-吲哚-4-基)丙-2-烯酸
式67
[步骤1]5-(3-甲基氧杂环丁烷-3-基)-3-(2,4,6-三氯苯基)-1,2-唑-4-甲酸五氟苯基酯
在冰冷却下,向参考实施例X-11获得的化合物(10.2g)的二氯甲烷溶液(100ml)中依次滴加N,N-二(丙烷-2-基)胺(7.6ml)和三氟乙酸五氟苯基酯(7.3ml),于室温下将混合物搅拌过夜。将反应液减压浓缩,将获得的残留物经硅胶柱色谱(正己烷/二氯甲烷)进行纯化,得到标题化合物(11.4g)。
1H-NMR(CDCl3)δ:1.92(3H,s),4.70(2H,d,J=6.7Hz),5.21(2H,d,J=6.7Hz),7.46(2H,s)。
[步骤2](2E)-3-(1-{[5-(3-甲基氧杂环丁烷-3-基)-3-(2,4,6-三氯苯基)-1,2-唑-4-基]羰基}-1H-吲哚-4-基)丙-2-烯酸叔丁基酯
在冰冷却下,向参考实施例E-1获得的化合物(4.98g)的N,N-二甲基甲酰胺溶液(60ml)中缓慢加入氢化钠(55%的油分散液,936mg),于室温下将混合物搅拌45分钟。在冰冷却下,向反应液中加入上述步骤1获得的化合物(10.3g)的N,N-二甲基甲酰胺溶液(50ml),于室温下将混合物搅拌30分钟。在冰冷却下,向反应液中加入水,用乙酸乙酯进行萃取。将萃取液用水和饱和盐水依次洗涤,经无水硫酸钠干燥。减压蒸除溶剂,将获得的残留物经硅胶柱色谱(正己烷/乙酸乙酯)进行纯化,得到标题化合物(11.9g)。
1H-NMR(CDCl3)δ:1.55(9H,s),1.92(3H,s),4.60(2H,d,J=6.7Hz),5.12(2H,d,J=6.7Hz),6.45(1H,d,J=16.3Hz),6.71(1H,d,J=3.6Hz),7.13(1H,d,J=4.2Hz),7.31(2H,s),7.36(1H,t,J=7.9Hz),7.53(1H,d,J=7.3Hz),7.85(1H,d,J=16.3Hz),8.29(1H,d,J=8.5Hz)。
[步骤3](2E)-3-(1-{[5-(3-甲基氧杂环丁烷-3-基)-3-(2,4,6-三氯苯基)-1,2-唑-4-基]羰基}-1H-吲哚-4-基)丙-2-烯酸
向上述步骤2获得的化合物(11.9g)的四氢呋喃溶液(20ml),加入甲酸(120ml),于室温下将混合物搅拌2小时。将反应液用二氯甲烷稀释,用水和饱和盐水依次洗涤、经无水硫酸钠干燥。减压蒸除溶剂,将获得的残留物经硅胶柱色谱(二氯甲烷/甲醇)进行纯化。将溶离部份在减压浓缩,于获得的残留物中加入正己烷/二氯甲烷混合液,经滤取、干燥后得到标题化合物(7.61g)。
1H-NMR(CDCl3)δ:1.93(3H,s),4.62(2H,d,J=6.7Hz),5.13(2H,d,J=6.7Hz),6.55(1H,d,J=15.7Hz),6.74(1H,d,J=4.2Hz),7.17(1H,d,J=4.2Hz),7.32(2H,s),7.40(1H,t,J=7.3Hz),7.59(1H,d,J=7.3Hz),8.05(1H,d,J=15.7Hz),8.34(1H,d,J=7.9Hz)。
MS(m/z):531(M+H)+。
使用在参考实施例中获得的化合物,通过与实施例1相同的方法,得到下述化合物。
[表30]
[表31]
[表32]
[实施例10](2E)-3-(3-甲基-1-{[5-(1-甲基环丁基)-3-(2,4,6-三氯苯基)-1,2-唑-4-基]羰基}-1H-吲哚-4-基)丙-2-烯酸
式68
[步骤1]5-(1-甲基环丁基)-3-(2,4,6-三氯苯基)-1,2-唑-4-甲酸五氟苯基酯
使用参考实施例X-14获得的化合物(0.109g),通过与实施例1步骤1相同的方法,得到标题化合物(0.123g)。
1H-NMR(CDCl3)δ:1.72(3H,s),1.92-2.01(1H,m),2.14-2.30(3H,m),2.72-2.82(2H,m),7.44(2H,s)。
MS(m/z):526(M+H)+。
[步骤2](2E)-3-(3-甲基-1-{[5-(1-甲基环丁基)-3-(2,4,6-三氯苯基)-1,2-唑-4-基]羰基}-1H-吲哚-4-基)丙-2-烯酸叔丁基酯
使用上述步骤1获得的化合物(0.123g)和参考实施例E-8获得的化合物(0.054g),通过与实施例1步骤2相同的方法,得到标题化合物(0.103g)。
1H-NMR(CDCl3)δ:1.54(9H,s),1.67(3H,s),1.88-2.22(4H,m),2.38(3H,s),2.56-2.63(2H,m),6.36(1H,d,J=15.7Hz),6.95(1H,s),7.29-7.33(3H,m),7.52(1H,d,J=7.9Hz),8.28(1H,d,J=15.7Hz),8.37(1H,d,J=8.5Hz)。
MS(m/z):599(M+H)+。
[步骤3](2E)-3-(3-甲基-1-{[5-(1-甲基环丁基)-3-(2,4,6-三氯苯基)-1,2-唑-4-基]羰基}-1H-吲哚-4-基)丙-2-烯酸
在冰冷却下,向上述步骤2获得的化合物(0.100g)的二氯甲烷(5ml)溶液中加入三氟乙酸(1ml),于室温下将混合物搅拌过夜。将反应液减压浓缩,向获得的残留物中加入乙酸乙酯和水,分离两层。将有机层用水洗涤后,经无水硫酸钠干燥。经减压蒸除溶剂后得到标题化合物(0.072g)。
1H-NMR(CDCl3)δ:1.68(3H,s),1.88-2.17(4H,m),2.39(3H,s),2.56-2.64(2H,m),6.46(1H,d,J=15.7Hz),6.98(1H,s),7.32(2H,s),7.35(1H,t,J=7.9Hz),7.58(1H,d,J=7.9Hz),8.41(1H,d,J=7.9Hz),8.48(1H,d,J=15.7Hz)。
MS(m/z):543(M+H)+。
使用参考实施例中获得的化合物,通过与实施例1相同的方法,得到下述化合物。
[表33]
[表34]
[表35]
[表36]
[实施例21](2E)-3-(1-{[5-(2-氟丙烷-2-基)-3-(2,4,6-三氯苯基)-1,2-唑-4-基]羰基}-3-甲基-1H-吲哚-4-基)丙-2-烯酸
式69
[步骤1](2E)-3-(1-{[5-(2-氟丙烷-2-基)-3-(2,4,6-三氯苯基)-1,2-唑-4-基]羰基}-3-甲基-1H-吲哚-4-基)丙-2-烯酸叔丁基酯
向参考实施例X-20获得的化合物(200mg)和N,N-二甲基甲酰胺(10μl)的二氯甲烷(6ml)溶液中加入草酰氯(216mg),于室温下将混合物搅拌30分钟。将反应液减压浓缩得到酰氯。
在冰冷却下,向参考实施例E-8获得的化合物(218mg)、N,N-二(丙烷-2-基)乙基胺(0.19ml)和4-二甲基氨基吡啶(7.0mg)的二氯甲烷(3ml)溶液中滴加上述酰氯的二氯甲烷(3ml)溶液,于室温下将混合物搅拌过夜。将反应液倾至正己烷、乙酸乙酯和水的混合液,分离两层。将有机层用水和饱和盐水依次洗涤,经无水硫酸钠干燥。减压蒸除溶剂,将获得的残留物经硅胶柱色谱(正己烷/乙酸乙酯)进行纯化,得到标题化合物(171mg)。
1H-NMR(CDCl3)δ:1.54(9H,s),1.88(6H,d,J=21.8Hz),2.41(3H,s),6.36(1H,d,J=15.7Hz),6.98(1H,s),7.31(1H,dd,J=8.2,4.1Hz),7.35(2H,s),7.53(1H,d,J=7.9Hz),8.30(1H,d,J=15.7Hz),8.41(1H,d,J=7.9Hz)。
MS(m/z):591(M+H)+。
[步骤2](2E)-3-(1-{[5-(2-氟丙烷-2-基)-3-(2,4,6-三氯苯基)-1,2-唑-4-基]羰基}-3-甲基-1H-吲哚-4-基)丙-2-烯酸
使用在上述步骤1获得的化合物(170mg),通过与实施例10的步骤3相同的方法,得到标题化合物(110mg)。
1H-NMR(CDCl3)δ:1.88(6H,d,J=21.8Hz),2.42(3H,d,J=1.2Hz),6.45(1H,d,J=15.7Hz),7.02(1H,s),7.35(3H,t,J=7.9Hz),7.58(1H,d,J=7.3Hz),8.48(2H,dd,J=13.0,6.5Hz)。
MS(m/z):535(M+H)+。
使用参考实施例中获得的化合物,通过与实施例21相同的方法,得到下述化合物。
[表37]
[表38]
[表39]
[表40]
[表41]
[表42]
[表43]
[表44]
[表45]
[表46]
[表47]
[表48]
[表49]
[表50]
[表51]
[表52]
[表53]
[实施例71](2E)-3-(3-环丙基-1-{[5-(2-氟丙烷-2-基)-3-(2,4,6-三氯苯基)-1,2-唑-4-基]羰基}-3-甲基-1H-吲哚-4-基)丙-2-烯酸
式70
[步骤1](2E)-3-(3-溴-1-{[5-(2-氟丙烷-2-基)-3-(2,4,6-三氯苯基)-1,2-唑-4-基]羰基}-3-甲基-1H-吲哚-4-基)丙-2-烯酸叔丁基酯
使用参考实施例X-20获得的化合物(2.08g)和参考实施例E-5获得的化合物(1.90g),通过与实施例21步骤1相同的方法,得到标题化合物(3.27g)。
1H-NMR(CDCl3)δ:1.55(9H,s),1.89(6H,d,J=21.8Hz),6.36(1H,d,J=15.7Hz),7.29-7.41(4H,m),7.57(1H,d,J=7.3Hz),8.43(1H,d,J=8.5Hz),8.88(1H,d,J=15.7Hz)。
[步骤2](2E)-3-(3-环丙基-1-{[5-(2-氟丙烷-2-基)-3-(2,4,6-三氯苯基)-1,2-唑-4-基]羰基}-3-甲基-1H-吲哚-4-基)丙-2-烯酸叔丁基酯
将上述步骤1获得的化合物(1.50g)、环丙基硼酸(0.216g)、磷酸三钾(1.45g)、三环己基膦(0.128g)和乙酸钯(51.3mg)悬浮于甲苯(30ml)和水(0.1ml)中,将混合物在氮环境下、于90℃搅拌2小时。冷却后,将反应液用乙酸乙酯稀释,用水和饱和盐水依次洗涤,经无水硫酸镁干燥。减压蒸除溶剂,将获得的残留物经硅胶柱色谱(正己烷/乙酸乙酯)进行纯化,得到标题化合物(863.9mg)。
1H-NMR(CDCl3)δ:0.54-0.59(2H,m),1.01(2H,dd,J=8.2,1.5Hz),1.54(9H,s),1.87(6H,d,J=21.2Hz),1.88-1.99(0H,m),6.38(1H,d,J=15.7Hz),6.87(1H,s),7.29-7.36(3H,m),7.56(1H,d,J=7.9Hz),8.40(1H,d,J=8.5Hz),8.74(1H,d,J=15.7Hz)。
[步骤3](2E)-3-(3-环丙基-1-{[5-(2-氟丙烷-2-基)-3-(2,4,6-三氯苯基)-1,2-唑-4-基]羰基}-3-甲基-1H-吲哚-4-基)丙-2-烯酸
使用在上述步骤2获得的化合物(863.9mg),通过与实施例10的步骤3相同的方法,得到标题化合物(547.0mg)。
1H-NMR(CDCl3)δ:0.55-0.60(2H,m),1.01-1.05(2H,m),1.87(6H,d,J=21.8Hz),1.88-1.97(7H,m),6.46(1H,d,J=15.7Hz),6.91(1H,s),7.33-7.38(3H,m),7.61(1H,d,J=7.9Hz),8.44(1H,d,J=8.5Hz),8.97(1H,d,J=15.7Hz)。
MS(m/z):559(M-H)-。
[实施例72](2E)-3-(1-{[5-(叔丁基)-3-(2,4,6-三氯苯基)-1,2-唑-4-基]羰基}-3-(二氟甲基)-1H-吲哚-4-基)丙-2-烯酸
式71
[步骤1](2E)-3-(1-{[5-(叔丁基)-3-(2,4,6-三氯苯基)-1,2-唑-4-基]羰基}-3-甲酰基-1H-吲哚-4-基)丙-2-烯酸叔丁基酯
使用参考实施例X-5获得的化合物(1.20g)和参考实施例E-6获得的化合物(0.93g),通过与实施例21步骤1相同的方法,得到标题化合物(1.95g)。
1H-NMR(CDCl3)δ:1.50(9H,s),1.58(9H,s),6.37(1H,d,J=16.3Hz),7.27-7.28(2H,m),7.46(1H,dd,J=8.2,4.1Hz),7.68(1H,d,J=7.3Hz),7.90(1H,s),8.41(1H,d,J=8.5Hz),8.88(1H,d,J=16.3Hz),9.96(1H,s)。
MS(m/z):545(M+H)+。
[步骤2](2E)-3-(1-{[5-(叔丁基)-3-(2,4,6-三氯苯基)-1,2-唑-4-基]羰基}-3-(二氟甲基)-1H-吲哚-4-基]丙-2-烯酸叔丁基酯
在氮环境中、冰冷却下,向上述步骤1获得的化合物(300mg)的二氯甲烷(5ml)溶液中加入二乙基氨基三氟化硫(0.36ml),将混合物在与上述相同的温度下搅拌4小时,于室温下搅拌过夜。将反应液倾至乙酸乙酯和饱和碳酸氢钠水的混合液中,分离两层。将有机层用水和饱和盐水依次洗涤,经无水硫酸钠干燥。减压蒸除溶剂,将获得的残留物经硅胶柱色谱(正己烷/乙酸乙酯)进行纯化,得到标题化合物(282mg)。
1H-NMR(CDCl3)δ:1.47(9H,s),1.55(9H,s),6.37(1H,d,J=15.1Hz),6.89(1H,t,J=55.0Hz),7.28-7.31(2H,m),7.43(1H,dd,J=7.9,3.9Hz),7.49(1H,dd,J=1.8,0.9Hz),7.58(1H,d,J=7.3Hz),8.06(1H,d,J=15.7Hz),8.43(1H,d,J=7.3Hz)。
MS(m/z):623(M+H)+。
[步骤3](2E)-3-(1-{[5-(叔丁基)-3-(2,4,6-三氯苯基)-1,2-唑-4-基]羰基}-3-(二氟甲基)-1H-吲哚-4-基)丙-2-烯酸
使用在上述步骤2获得的化合物(280mg),通过与实施例10的步骤3相同的方法,得到标题化合物(93mg)。
1H-NMR(CDCl3)δ:1.48(9H,s),6.47(1H,d,J=15.7Hz),6.89(1H,t,J=55.0Hz),7.30(2H,s),7.47(1H,dd,J=7.9,3.9Hz),7.51(1H,dd,J=2.1,1.1Hz),7.64(1H,d,J=7.9Hz),8.27(1H,d,J=15.7Hz),8.48(1H,d,J=7.9Hz)。
MS(m/z):567(M+H)+。
[实施例73](2E)-3-(1-{[5-(6-乙烯基吡啶-3-基)-3-(2,4,6-三氯苯基)-1,2-唑-4-基]羰基}-1H-吲哚-4-基)丙-2-烯酸
式72
[步骤1](2E)-3-(1-{[5-(6-氯吡啶-3-基)-3-(2,4,6-三氯苯基)-1,2-唑-4-基]羰基}-1H-吲哚-4-基)丙-2-酸叔丁基酯
使用参考实施例X-54获得的化合物(464mg)和参考实施例E-1获得的化合物(0.31g),通过与实施例21步骤1相同的方法,得到标题化合物(518mg)。
1H-NMR(CDCl3)δ:1.54(9H,s),6.46(1H,d,J=15.7Hz),6.71-6.76(1H,m),7.14(1H,d,J=4.2Hz),7.38-7.40(4H,m),7.56(1H,d,J=7.9Hz),7.85(1H,d,J=15.7Hz),8.00(1H,dd,J=8.5,1.8Hz),8.39(1H,d,J=7.9Hz),8.77(1H,d,J=2.4Hz)。
MS(m/z):628(M+H)+。
[步骤2](2E)-3-(1-{[5-(6-乙烯基吡啶-3-基)-3-(2,4,6-三氯苯基)-1,2-唑-4-基]羰基}-1H-吲哚-4-基)丙-2-烯酸叔丁基酯
向上述步骤1获得的化合物(250mg)、乙烯基三氟硼酸钾(84mg)和N,N-二(丙烷-2-基)乙基胺(0.14ml)的(丙烷-2-基)醇(3ml)和水(0.6ml)悬浮液中,加入[1,1’-双(二苯基膦基)二茂铁]二氯化钯(II)-二氯甲烷加成物,将混合物使用微波装置,在120℃搅拌1小时。冷却后将反应液倾至乙酸乙酯和水的混合液中,分离两层。将有机层用水和饱和盐水依次洗涤,经无水硫酸钠干燥。减压蒸除溶剂,将获得的残留物经硅胶柱色谱(正己烷/乙酸乙酯)进行纯化,得到标题化合物(50mg)。
1H-NMR(CDCl3)δ:1.56(9H,s),5.61(1H,d,J=10.9Hz),6.30(1H,d,J=17.5Hz),6.46(1H,d,J=16.3Hz),6.72(1H,d,J=4.2Hz),6.79(1H,dd,J=17.2,10.6Hz),7.17(1H,d,J=3.6Hz),7.37(2H,dd,J=15.1,7.9Hz),7.41(2H,s),7.56(1H,d,J=7.9Hz),7.85(1H,d,J=15.7Hz),7.94(1H,dd,J=8.5,2.4Hz),8.42(1H,d,J=8.5Hz),8.92(1H,d,J=2.4Hz)。
MS(m/z):620(M+H)+。
[步骤3](2E)-3-(1-{[5-(6-乙烯基吡啶-3-基)-3-(2,4,6-三氯苯基)-1,2-唑-4-基]羰基}-1H-吲哚-4-基)丙-2-烯酸
使用在上述步骤2获得的化合物(49mg),通过与实施例10的步骤3相同的方法,得到标题化合物(5.5mg)。
1H-NMR(CDCl3)δ:5.62(1H,d,J=10.9Hz),6.30(1H,d,J=17.5Hz),6.51(1H,d,J=15.7Hz),6.75-6.82(2H,m),7.20(1H,d,J=3.6Hz),7.39-7.42(4H,m),7.60(1H,d,J=7.3Hz),7.99-8.02(2H,m),8.47(1H,d,J=7.9Hz),8.90(1H,s)。
MS(m/z):564(M+H)+。
[实施例74](2E)-3-(1-{[5-(1-甲氧基环丁基)-3-(2,4,6-三氯苯基)-1,2-唑-4-基]羰基}-1H-吲哚-4-基)丙-2-烯酸
式73
[步骤1](2E)-3-(1-{[5-(1-甲氧基环丁基)-3-(2,4,6-三氯苯基)-1,2-唑-4-基]羰基}-1H-吲哚-4-基)丙-2-烯酸叔丁基酯
使用参考实施例X-15获得的化合物(0.316g)和参考实施例E-1获得的化合物(0.204g),通过与实施例21步骤1相同的方法,得到标题化合物(316mg),直接用于下一反应。
[步骤2](2E)-3-(1-{[5-(1-甲氧基环丁基)-3-(2,4,6-三氯苯基)-1,2-唑-4-基]羰基}-1H-吲哚-4-基)丙-2-烯酸
使用在上述步骤1获得的化合物(316mg),通过与实施例1步骤3相同的方法,得到标题化合物(0.147g)。
1H-NMR(CDCl3)δ:1.97-2.10(2H,m),2.42-2.50(2H,m),2.65-2.72(2H,m),3.13(3H,s),6.57(1H,d,J=15.7Hz),6.86(1H,d,J=4.2Hz),7.28(1H,d,J=4.2Hz),7.35-7.39(3H,m),7.58(1H,d,J=7.3Hz),8.11(1H,d,J=15.7Hz),8.44(1H,d,J=8.5Hz)。
使用参考实施例中获得的化合物,通过与实施例74相同的方法,得到下述化合物。
[表54]
[表55]
[表56]
[实施例81](2E)-3-(1-{[5-[1-(二甲基氨基)环丁基]-3-(2,4,6-三氯苯基)-1,2-唑-4-基]羰基}-3-甲基-1H-吲哚-4-基)丙-2-烯酸
式74
[步骤1]5-[1-(二甲基氨基)环丁基]-3-(2,4,6-三氯苯基)-1,2-唑-4-甲酸五氟苯基酯
使用参考实施例X-60获得的化合物(0.050g),通过与实施例1步骤1相同的方法,得到标题化合物(0.061g)。
1H-NMR(CDCl3)δ:1.85-1.94(1H,m),2.04-2.18(1H,m),2.21(6H,s),2.45-2.53(2H,m),2.74-2.82(2H,m),7.45(2H,s)。
[步骤2](2E)-3-(1-{[5-[1-(二甲基氨基)环丁基]-3-(2,4,6-三氯苯基)-1,2-唑-4-基]羰基}-3-甲基-1H-吲哚-4-基)丙-2-烯酸叔丁基酯
使用上述步骤1获得的化合物(416mg)和参考实施例E-8获得的化合物(0.174g),通过与实施例1步骤2相同的方法,得到标题化合物(0.283g)。
1H-NMR(CDCl3)δ:1.54(9H,s),1.83-1.95(2H,m),2.20(6H,s),2.38(3H,s),2.40-2.47(2H,m),2.51-2.61(2H,m),6.36(1H,d,J=15.7Hz),7.00(1H,s),7.27-7.33(3H,m),7.52(1H,d,J=7.3Hz),8.28(1H,d,J=15.7Hz),8.39(1H,d,J=7.9Hz)。
[步骤3](2E)-3-(1-{[5-[1-(二甲基氨基)环丁基]-3-(2,4,6-三氯苯基)-1,2-唑-4-基]羰基}-3-甲基-1H-吲哚-4-基)丙-2-烯酸
向上述步骤2获得的化合物(0.283g)的二氯甲烷(3ml)溶液中加入4N盐酸的1,4-二烷溶液(1ml),于室温下将混合物搅拌1.5小时。再向反应液中加入4N盐酸的1,4-二烷溶液(3ml),进一步搅拌1小时。将反应液减压浓缩,向获得的残留物中加入水和二氯甲烷,分离两层后,将有机层经无水硫酸钠干燥。减压蒸除溶剂,将获得的残留物经硅胶柱色谱(正己烷/乙酸乙酯)进行纯化。将洗脱的组分减压浓缩,向获得的残留物中加入正己烷/乙醚混合液,过滤收集产生的固体,干燥后得到标题化合物(0.161g)。
1H-NMR(CDCl3)δ:1.84-1.97(2H,m),2.21(6H,s),2.38(3H,s),2.40-2.48(2H,m),2.52-2.61(2H,m),6.45(1H,d,J=15.7Hz),7.02(1H,s),7.31-7.36(3H,m),7.56(1H,d,J=7.9Hz),8.42-8.47(2H,m)。
MS(m/z):572(M+H)+。
使用参考实施例中获得的化合物,通过与实施例81相同的方法,得到下述化合物。
[表57]
[实施例83](2E)-3-(3-甲基-1-{[5-[1-(甲基氨基)环丁基]-3-(2,4,6-三氯苯基)-1,2-唑-4-基]羰基}-1H-吲哚-4-基)丙-2-烯酸
式75
[步骤1]5-(1-{甲基[(2-硝基苯基)磺酰基]氨基}环丁基)-3-(2,4,6-三氯苯基)-1,2-唑-4-甲酸五氟苯基酯
使用参考实施例X-62获得的化合物,通过与实施例1步骤1相同的方法,得到标题化合物(0.454g)。
1H-NMR(CDCl3)δ:1.79-1.91(1H,m),1.94-2.03(1H,m),2.86-3.01(4H,m),3.09(3H,s),7.46(2H,s),7.59-7.72(3H,m),7.90(1H,dd,J=7.9,1.2Hz)。
[步骤2](2E)-3-(3-甲基-1-{[5-(1-{甲基[(2-硝基苯基)磺酰基]氨基}环丁基)-3-(2,4,6-三氯苯基)-1,2-唑-4-基]羰基}-1H-吲哚-4-基)丙-2-烯酸叔丁基酯
使用上述步骤1获得的化合物(0.447g)和参考实施例E-8获得的化合物(0.158g),通过与实施例1步骤2相同的方法,得到标题化合物(0.386g)。
1H-NMR(CDCl3)δ:1.54(9H,s),1.83-2.01(2H,m),2.34(3H,d,J=1.2Hz),2.63-2.72(2H,m),2.80-2.87(2H,m),3.05(3H,s),6.35(1H,d,J=15.7Hz),7.04(1H,d,J=1.2Hz),7.26-7.32(3H,m),7.51(1H,d,J=7.3Hz),7.60-7.71(3H,m),7.96-7.99(1H,m),8.26(1H,d,J=15.7Hz),8.37(1H,d,J=8.5Hz)。
MS(m/z):799(M+H)+。
[步骤3](2E)-3-[3-甲基-1-({5-[1-(甲基氨基)环丁基]-3-(2,4,6-三氯苯基)-1,2-唑-4-基}羰基)-1H-吲哚-4-基]丙-2-烯酸叔丁基酯
在冰冷却下,向上述步骤2获得的化合物(0.376g)的乙腈(10ml)溶液中加入4-溴苯硫醇(0.192g)和碳酸钾(0.281g),将混合物在与上述相同的温度下搅拌1小时,于室温下搅拌3小时。在冰冷却下,向反应液中加入乙酸乙酯和水,分离两层。将有机层用饱和盐水洗涤,经无水硫酸钠干燥。减压蒸除溶剂,将获得的残留物经硅胶柱色谱(正己烷/乙酸乙酯)进行纯化,得到标题化合物(0.244g)。
1H-NMR(CDCl3)δ:1.54(9H,s),1.97-2.28(4H,m),2.25(3H,s),2.34(3H,d,J=1.2Hz),2.60-2.67(2H,m),6.36(1H,d,J=15.7Hz),6.93(1H,d,J=1.2Hz),7.29-7.33(3H,m),7.52(1H,d,J=7.9Hz),8.26(1H,d,J=15.7Hz),8.35(1H,d,J=8.5Hz)。
MS(m/z):614(M+H)+。
[步骤4](2E)-3-[3-甲基-1-({5-[1-(甲基氨基)环丁基]-3-(2,4,6-三氯苯基)-1,2-唑-4-基}羰基)-1H-吲哚-4-基]丙-2-烯酸
使用上述步骤3获得的化合物(0.100g),通过与实施例10的步骤3相同的方法,得到标题化合物(0.085g)。
1H-NMR(CDCl3)δ:1.99-2.39(4H,m),2.30(3H,s),2.33(3H,s),2.62-2.70(2H,m),6.43(1H,d,J=15.7Hz),6.94(1H,s),7.31(2H,s),7.35(1H,t,J=7.9Hz),7.55(1H,d,J=7.9Hz),8.37-8.43(2H,m)。
MS(ESI):558(M+H)+。
使用在参考实施例中获得的化合物,通过与实施例83相同的方法,得到下述化合物。
[表58]
[表59]
[实施例87](2E)-3-(1-{[5-{1-[丙烯酰基(甲基)氨基]环丁基}-3-(2,4,6-三氯苯基)-1,2-唑-4-基]羰基}-3-甲基-1H-吲哚-4-基)丙-2-烯酸
式76
[步骤1](2E)-3-(1-{[5-{1-[丙烯酰基(甲基)氨基]环丁基}-3-(2,4,6-三氯苯基)-1,2-唑-4-基]羰基}-3-甲基-1H-吲哚-4-基)丙-2-烯酸叔丁基酯
向实施例83的步骤3获得的化合物(0.133g)的二氯甲烷(3ml)溶液中加入三乙基胺(0.090ml)和丙烯酰氯(0.035ml),于室温下将混合物搅拌1小时。向反应液加入二氯甲烷和水,分离两层后,将有机层经无水硫酸钠干燥。经减压蒸除溶剂后得到标题化合物,将此直接用于下一反应。
MS(m/z):668(M+H)+。
[步骤2](2E)-3-(1-{[5-{1-[丙烯酰基(甲基)氨基]环丁基}-3-(2,4,6-三氯苯基)-1,2-唑-4-基]羰基}-3-甲基-1H-吲哚-4-基)丙-2-烯酸
使用上述步骤1获得的化合物,通过与实施例10的步骤3相同的方法,得到标题化合物(0.112g)。
1H-NMR(CDCl3)δ:1.94-2.02(1H,m),2.32-2.42(1H,m),2.34(3H,s),2.53-3.09(4H,m),3.04(3H,s),5.32(1H,d,J=10.3Hz),5.83(1H,d,J=16.9Hz),6.16(1H,dd,J=16.9,10.3Hz),6.41(1H,d,J=15.7Hz),6.95(1H,s),7.26-7.32(3H,m),7.53(1H,d,J=7.9Hz),8.40(1H,d,J=7.9Hz),8.44(1H,d,J=15.7Hz)。
MS(m/z):612(M+H)+。
[实施例88](2E)-3-(1-{[5-{1-[(二甲基氨基)甲基]环丁基}-3-(2,4,6-三氯苯基)-1,2-唑-4-基]羰基}-3-甲基-1H-吲哚-4-基)丙-2-烯酸
式77
[步骤1]5-{1-[(甲氧基甲氧基)甲基]环丁基}-3-(2,4,6-三氯苯基)-1,2-唑-4-甲酸五氟苯基酯
使用参考实施例X-33获得的化合物(2.04g),通过与实施例1步骤1相同的方法,得到标题化合物(2.34g)。
1H-NMR(CDCl3)δ:1.98-2.11(1H,m),2.14-2.22(1H,m),2.41-2.48(2H,m),2.73-2.83(2H,m),3.24(3H,s),4.03(2H,s),4.59(2H,s),7.44(2H,s)。
MS(m/z):586(M+H)+。
[步骤2](2E)-3-(1-{[5-{1-[(甲氧基甲氧基)甲基]环丁基}-3-(2,4,6-三氯苯基)-1,2-唑-4-基]羰基}-3-甲基-1H-吲哚-4-基)丙-2-烯酸叔丁基酯
使用在上述步骤1获得的化合物(2.34g)和参考实施例E-8获得的化合物(1.03g),通过与实施例1步骤2相同的方法,得到标题化合物(2.00g)。
1H-NMR(CDCl3)δ:1.54(9H,s),1.95-2.24(4H,m),2.36(3H,s),2.59-2.66(2H,m),3.23(3H,s),4.01(2H,s),4.50(2H,s),6.36(1H,d,J=15.7Hz),7.09(1H,d,J=1.2Hz),7.28-7.32(1H,m),7.31(2H,s),7.52(1H,d,J=7.9Hz),8.28(1H,d,J=15.7Hz),8.38(1H,d,J=8.5Hz)。
MS(m/z):659(M+H)+。
[步骤3](2E)-3-[1-({5-[1-(羟基甲基)环丁基]-3-(2,4,6-三氯苯基)-1,2-唑-4-基}羰基)-3-甲基-1H-吲哚-4-基]丙-2-烯酸
向上述步骤2获得的化合物(1.47g)的二氯甲烷(10ml)溶液中加入三氟乙酸(2ml),于室温下将混合物搅拌1.5小时。再向反应液中加入三氟乙酸(1ml)并继续搅拌30分钟,再追加三氟乙酸(2ml)并搅拌30分钟。向反应液中加入二氯甲烷和水,分离两层。将有机层用水和饱和盐水依次洗涤,经无水硫酸钠干燥。减压蒸除溶剂,向获得的残留物中加入正己烷,过滤收集产生的固体,得到标题化合物(1.32g)。
MS(m/z):559(M+H)+。
[步骤4](2E)-3-(1-{[5-(1-甲酰基环丁基)-3-(2,4,6-三氯苯基)-1,2-唑-4-基]羰基}-3-甲基-1H-吲哚-4-基)丙-2-烯酸
向上述步骤3获得的化合物(0.60g)的二氯甲烷(5ml)溶液加入Dess-Martin过碘酸(0.909g),于室温下将混合物搅拌2小时。向反应液中加入水和二氯甲烷,分离为两层。将有机层用水、硫代硫酸钠水溶液和饱和盐水依次洗涤,经无水硫酸钠干燥。减压蒸除溶剂,向获得的残留物中加入乙醚和正己烷,过滤收集产生的固体,得到标题化合物(0.460g)。
MS(m/z):557(M+H)+。
[步骤5](2E)-3-(1-{[5-{1-[(二甲基氨基)甲基]环丁基}-3-(2,4,6-三氯苯基)-1,2-唑-4-基]羰基}-3-甲基-1H-吲哚-4-基)丙-2-烯酸
向上述步骤4获得的化合物(0.160g)的1,2-二氯乙烷(3ml)悬浮液中加入二甲基胺(2.0M的四氢呋喃溶液,0.57ml)和乙酰氧基硼氢化钠(0.304g),于室温下将混合物搅拌2小时。向反应液中加入二氯甲烷和水,分离两层。将水层用二氯甲烷进行萃取,合并有机层,用水洗涤后,经无水硫酸钠干燥。减压蒸除溶剂,将获得的残留物经硅胶柱色谱(正己烷/乙酸乙酯)进行纯化,以制备型薄层色谱(乙酸乙酯)进行纯化。向获得的残留物中加入正己烷,过滤收集产生的固体,得到标题化合物(0.028g)。
1H-NMR(CDCl3)δ:2.01(6H,s),2.05-2.30(4H,m),2.35(3H,d,J=1.2Hz),2.69-2.76(2H,m),2.89(2H,s),6.40(1H,d,J=15.7Hz),7.04(1H,d,J=1.2Hz),7.27(2H,s),7.31(1H,t,J=7.9Hz),7.52(1H,d,J=7.9Hz),8.39-8.45(2H,m)。
MS(m/z):586(M+H)+。
[实施例89](2E)-3-(1-{[5-[1-(二甲基氨基甲酰基)环丁基]-3-(2,4,6-三氯苯基)-1,2-唑-4-基]羰基}-3-甲基-1H-吲哚-4-基)丙-2-烯酸
式78
[步骤1](2E)-3-[1-({5-[1-(羟基甲基)环丁基]-3-(2,4,6-三氯苯基)-1,2-唑-4-基}羰基)-3-甲基-1H-吲哚-4-基)丙-2-烯酸2,2,2-三氯乙基酯
向实施例88的步骤3获得的化合物(0.398g)的2,2,2-三氯乙醇(3.5ml)溶液中加入1-乙基-3-(3-二甲基氨基丙基)-碳二亚胺盐酸盐(0.204g)和1-羟基苯并三唑一水合物(0.035g),于室温下将混合物搅拌1小时后,在70℃搅拌1.5小时。向反应液中加入乙酸乙酯和水,分离两层。将有机层用饱和盐水洗涤,经无水硫酸钠干燥。减压蒸除溶剂,将获得的残留物经硅胶柱色谱(正己烷/乙酸乙酯)进行纯化,得到标题化合物(0.262g)。
1H-NMR(CDCl3)δ:1.97-2.06(1H,m),2.11-2.23(1H,m),2.27-2.34(2H,m),2.32(3H,d,J=1.2Hz),2.51-2.59(2H,m),4.11(2H,d,J=5.4Hz),4.88(2H,s),6.52(1H,d,J=15.7Hz),7.01(1H,d,J=1.2Hz),7.29(2H,s),7.36(1H,t,J=7.9Hz),7.59(1H,d,J=7.9Hz),8.40(1H,d,J=7.9Hz),8.51(1H,d,J=15.7Hz)。
MS(m/z):689(M+H)+。
[步骤2](2E)-3-(1-{[5-(1-甲酰基环丁基)-3-(2,4,6-三氯苯基)-1,2-唑-4-基]羰基}-3-甲基-1H-吲哚-4-基)丙-2-烯酸2,2,2-三氯乙基酯
使用在上述步骤1获得的化合物(0.262g),通过与实施例88的步骤4相同的方法,得到标题化合物(0.261g)。
MS(m/z):687(M+H)+。
[步骤3]1-[4-({3-甲基-4-[(1E)-3-氧代-3-(2,2,2-三氯乙氧基)丙-1-烯-1-基]吲哚-1-羰基}-3-(2,4,6-三氯苯基)-1,2-唑5-基)环丁烷羧酸
向上述步骤2获得的化合物(0.261g)中加入叔丁醇(3ml)、乙腈(5ml)、水(1ml)、磷酸二氢钠二水合物(0.065g)、2-甲基-2-丁烯(0.18ml)和亚氯酸钠(0.077g),于室温下将混合物搅拌1.5小时。向反应液加入1N盐酸使其呈弱酸性,用二氯甲烷进行萃取。将萃取液用饱和盐水洗涤,经无水硫酸钠干燥。经减压蒸除溶剂后得到标题化合物(0.198g)。
1H-NMR(CDCl3)δ:1.99-2.38(2H,m),2.33(3H,s),2.73-2.79(4H,m),4.87(2H,s),6.51(1H,d,J=15.7Hz),7.00(1H,s),7.29-7.33(3H,m),7.57(1H,d,J=7.9Hz),8.35(1H,d,J=7.9Hz),8.52(1H,d,J=15.7Hz)。
MS(m/z):703(M+H)+。
[步骤4](1-{[5-[1-(二甲基氨基甲酰基)环丁基]-3-(2,4,6-三氯苯基)-1,2-唑-4-基]羰基}-3-甲基-1H-吲哚-4-基)丙-2-烯酸2,2,2-三氯乙基酯
向上述步骤3获得的化合物(0.064g)和O-(7-氧杂苯并三唑-1-基)-N,N,N’,N’-四甲基脲六氟磷酸盐(0.045g)的N,N-二甲基甲酰胺(1ml)溶液中加入二甲基胺(2.0M的四氢呋喃溶液,0.091ml),于室温下将混合物搅拌1小时。向反应液加入乙酸乙酯和饱和盐水,分离两层。将水层用乙酸乙酯进行萃取,合并有机层,用饱和盐水洗涤,经无水硫酸钠干燥。减压蒸除溶剂,将获得的残留物经硅胶柱色谱(正己烷/乙酸乙酯)进行纯化,得到标题化合物(0.068g)。
1H-NMR(CDCl3)δ:2.03-2.24(2H,m),2.43(3H,s),2.61(3H,s),2.65(3H,s),2.82-2.92(4H,m),4.88(2H,s),6.51(1H,d,J=15.7Hz),6.79(1H,d,J=1.2Hz),7.30(1H,t,J=7.9Hz),7.37(2H,s),7.58(1H,d,J=7.9Hz),8.36(1H,d,J=7.9Hz),8.58(1H,d,J=15.7Hz)。
MS(m/z):730(M+H)+。
[步骤5](2E)-3-(1-{[5-[1-(二甲基氨基甲酰基)环丁基]-3-(2,4,6-三氯苯基)-1,2-唑-4-基]羰基}-3-甲基-1H-吲哚-4-基)丙-2-烯酸
向上述步骤4获得的化合物(0.054g)的四氢呋喃(2ml)溶液中加入水(0.2ml)、铟(0.127g)和甲酸(0.08ml),将混合物使用微波装置,在100℃搅拌1小时。再向反应液中加入铟(0.085g),将混合物使用微波装置,在100℃搅拌30分钟。滤除无机物,向滤液中加入水和乙酸乙酯,分离两层。将有机层用饱和盐水洗涤,经无水硫酸钠干燥。减压蒸除溶剂,将获得的残留物经硅胶柱色谱(二氯甲烷/甲醇)进行纯化,用制备性薄层色谱(二氯甲烷/甲醇)进行纯化,得到标题化合物(0.009g)。
1H-NMR(CDCl3)δ:2.03-2.14(1H,m),2.17-2.28(1H,m),2.43(3H,s),2.62(3H,s),2.67(3H,s),2.83-2.96(4H,m),6.36(1H,d,J=15.7Hz),6.78(1H,s),7.29(1H,t,J=8.0Hz),7.38(2H,s),7.54(1H,d,J=8.0Hz),8.34(1H,d,J=8.0Hz),8.44(1H,d,J=15.7Hz)。
MS(m/z):600(M+H)+。
[实施例90](2E)-3-(1-{[5-(1-氨基环丁基)-3-(2,4,6-三氯苯基)-1,2-唑-4-基]羰基}-3-甲基-1H-吲哚-4-基)丙-2-烯酸
式79
[步骤1]5-{1-[(叔丁氧基羰基)(丙-2-烯-1-基)氨基]环丁基}-3-(2,4,6-三氯苯基)-1,2-唑-4-甲酸五氟苯基酯
使用参考实施例X-65获得的化合物,通过与实施例1步骤1相同的方法,得到标题化合物(1.01g)。
1H-NMR(CDCl3)δ:1.36(9H,s),1.91-2.06(2H,m),2.69-2.78(2H,m),2.88-2.96(2H,m),4.07-4.13(2H,m),5.19-5.28(2H,m),5.92-6.02(1H,m),7.45(2H,s)。
[步骤2](2E)-3-(1-{[5-{1-[(叔丁氧基羰基)(丙-2-烯-1-基)氨基]环丁基}-3-(2,4,6-三氯苯基)-1,2-唑-4-基]羰基}-3-甲基-1H-吲哚-4-基)丙-2-烯酸叔丁基酯
使用在上述步骤1获得的化合物(1.01g)和参考实施例E-8获得的化合物(0.389g),通过与实施例1步骤2相同的方法,得到标题化合物(0.899g)。
1H-NMR(CDCl3)δ:1.28(9H,brs),1.54(9H,s),1.89-1.99(1H,m),2.12-2.27(1H,m),2.31(3H,d,J=1.2Hz),2.60-2.79(2H,m),2.81-2.99(2H,m),3.60-3.74(2H,m),5.11-5.18(2H,m),5.79-5.89(1H,m),6.35(1H,d,J=15.7Hz),6.91(1H,brs),7.26-7.32(3H,m),7.51(1H,d,J=7.9Hz),8.25(1H,d,J=15.7Hz),8.39(1H,d,J=7.9Hz)。
MS(m/z):740(M+H)+。
[步骤3](2E)-3-[3-甲基-1-({5-[1-(丙-2-烯-1-基)环丁基]-3-(2,4,6-三氯苯基)-1,2-唑-4-基}羰基)-1H-吲哚-4-基)丙-2-烯酸
向上述步骤2获得的化合物(0.40g)的二氯甲烷(2ml)溶液加入4N盐酸的1,4-二烷(4ml)溶液,于室温下将混合物搅拌2.5小时。将反应液减压浓缩,向获得的残留物中加入三氟乙酸(3ml),于室温下将混合物搅拌1小时。将反应液减压浓缩,向获得的残留物中加入二氯甲烷和水,分离两层。将有机层用水洗涤后,经无水硫酸钠干燥。减压蒸除溶剂,向获得的残留物中加入乙醚和正己烷,过滤收集产生的固体,得到标题化合物(0.281g)。
1H-NMR(CDCl3)δ:1.99-2.10(1H,m),2.12-2.22(1H,m),2.27-2.36(2H,m),2.35(3H,s),2.63-2.70(2H,m),3.11(2H,d,J=6.0Hz),4.93-4.97(1H,m),5.00-5.06(1H,m),5.69-5.79(1H,m),6.45(1H,d,J=15.7Hz),6.95(1H,d,J=1.2Hz),7.32-7.37(3H,m),7.57(1H,d,J=7.9Hz),8.40(1H,d,J=7.9Hz),8.46(1H,d,J=15.7Hz)。
MS(m/z):584(M+H)+。
[步骤4](2E)-3-(1-{[5-(1-氨基环丁基)-3-(2,4,6-三氯苯基)-1,2-唑-4-基]羰基}-3-甲基-1H-吲哚-4-基)丙-2-烯酸
向上述步骤3获得的化合物(0.237g)、3-二苯基磷烷基(phosphanyl)丙基(二苯基)膦(0.084g)和双[(2,2,2-三氟乙酰)氧基]钯(0.067g)中加入乙腈(2ml)和水(0.2ml),在氮环境下,将混合物使用微波装置在110℃搅拌1小时。将不溶物滤去后,将滤液减压浓缩,将获得的残留物经硅胶柱色谱(二氯甲烷/甲醇)进行纯化,用制备性薄层色谱(二氯甲烷/甲醇)进行纯化。向获得的残留物中加入二氯甲烷,过滤收集产生的固体,得到标题化合物(0.020g)。
1H-NMR(DMSO-d6)δ:1.84-1.92(1H,m),2.01-2.10(3H,m),2.35(3H,s),2.65-2.70(2H,m),6.50(1H,d,J=15.7Hz),7.29(1H,s),7.33(1H,t,J=7.9Hz),7.68(1H,d,J=7.9Hz),7.83(2H,s),8.23-8.28(2H,m)。
MS(m/z):544(M+H)+。
[实施例91](2E)-3-[1-({5-[2-(二甲基氨基)丙烷-2-基]-3-(2,4,6-三氯苯基)-1,2-唑-4-基}羰基)-3-甲基-1H-吲哚-4-基]丙-2-烯酸
式80
[步骤1](2E)-3-[1-({5-[2-(二甲基氨基)丙烷-2-基]-3-(2,4,6-三氯苯基)-1,2-唑-4-基}羰基)-3-甲基-1H-吲哚-4-基]丙-2-烯酸叔丁基酯
向实施例84的步骤3获得的化合物(0.220g)的1,2-二氯乙烷(3ml)溶液加入甲醛(37%水溶液,0.136ml)和三乙酰氧基硼氢化钠(0.387g),于室温下将混合物搅拌1小时。向反应液中加入水和二氯甲烷,分离两层后,将有机层经无水硫酸钠干燥。经减压蒸除溶剂后得到标题化合物(0.199g)。
1H-NMR(CDCl3)δ:1.49(6H,brs),1.54(9H,s),2.14(6H,s),2.41(3H,d,J=1.2Hz),6.36(1H,d,J=15.7Hz),7.01(1H,s),7.30(1H,t,J=7.9Hz),7.34(2H,s),7.52(1H,d,J=7.9Hz),8.31(1H,d,J=15.7Hz),8.43(1H,d,J=7.9Hz)。
MS(m/z):616(M+H)+。
[步骤2](2E)-3-[1-({5-[2-(二甲基氨基)丙烷-2-基]-3-(2,4,6-三氯苯基)-1,2-唑-4-基}羰基)-3-甲基-1H-吲哚-4-基]丙-2-烯酸
使用在上述步骤1获得的化合物(0.199g),通过与实施例10的步骤3相同的方法,得到标题化合物(0.127g)。
1H-NMR(CDCl3)δ:1.51(6H,brs),2.15(6H,s),2.41(3H,s),6.42(1H,d,J=15.7Hz),7.03(1H,s),7.31-7.35(3H,m),7.56(1H,d,J=7.9Hz),8.46-8.50(2H,m)。
MS(m/z):516(M+H)+。
使用实施例86的步骤3获得的化合物,通过与实施例91相同的方法,得到下述化合物。
[表60]
[实施例93](2E)-3-(1-{[5-(1-氰基环丁基)-3-(2,4,6-三氯苯基)-1,2-唑-4-基]羰基}-3-甲基-1H-吲哚-4-基)丙-2-烯酸
式81
[步骤1](2E)-3-(1-{[5-(1-氨基甲酰基环丁基)-3-(2,4,6-三氯苯基)-1,2-唑-4-基]羰基}-3-甲基-1H-吲哚-4-基)丙-2-烯酸2,2,2-三氯乙基酯
向实施例89的步骤3获得的化合物(0.067g)和O-(7-氧杂苯并三唑-1-基)-N,N,N’,N’-四甲基脲六氟磷酸盐(0.047g)的N,N-二甲基甲酰胺(1ml)溶液中加入N,N-二(丙烷-2-基)乙基胺(0.049ml),于室温下将混合物搅拌5分钟,然后加入氯化铵(0.008g),于室温下将混合物搅拌过夜。向反应液中加入水和乙酸乙酯,分离两层。将水层用乙酸乙酯进行萃取,合并有机层,用饱和盐水洗涤,经无水硫酸钠干燥。减压蒸除溶剂,将获得的残留物经硅胶柱色谱(正己烷/乙酸乙酯)进行纯化,得到标题化合物(0.034g)。
1H-NMR(CDCl3)δ:1.90-2.00(1H,m),2.17-2.25(1H,m),2.33(3H,d,J=1.2Hz),2.55-2.63(2H,m),2.90-3.00(2H,m),4.88(2H,s),5.73(1H,brs),6.53(1H,d,J=15.7Hz),6.98(1H,d,J=1.2Hz),7.30(2H,s),7.38(1H,t,J=7.9Hz),7.59-7.64(2H,m),8.39(1H,d,J=7.9Hz),8.50(1H,d,J=15.7Hz)。
[步骤2](2E)-3-(1-{[5-(1-氰基环丁基)-3-(2,4,6-三氯苯基)-1,2-唑-4-基]羰基}-3-甲基-1H-吲哚-4-基)丙-2-烯酸2,2,2-三氯乙基酯
向在上述步骤1获得的化合物(0.290g)的吡啶(1ml)溶液中加入三氟乙酸酐(0.172ml),于室温下将混合物搅拌1小时。将反应液减压浓缩,将获得的残留物用乙酸乙酯稀释,用1N盐酸水溶液-饱和盐水(1:1)和饱和碳酸氢钠水溶液-饱和盐水(1:1)依次洗涤,经无水硫酸钠干燥。经减压蒸除溶剂后得到标题化合物(0.308g)。
1H-NMR(CDCl3)δ:2.15-2.25(1H,m),2.41(3H,s),2.44-2.53(1H,m),2.75-2.82(2H,m),2.92-2.99(2H,m),4.88(2H,s),6.53(1H,d,J=15.7Hz),6.95(1H,s),7.35-7.39(3H,m),7.61(1H,d,J=7.9Hz),8.41(1H,d,J=7.9Hz),8.55(1H,d,J=15.7Hz)。
MS(m/z):684(M+H)+。
[步骤3](2E)-3-(1-{[5-(1-氰基环丁基)-3-(2,4,6-三氯苯基)-1,2-唑-4-基]羰基}-3-甲基-1H-吲哚-4-基)丙-2-烯酸
使用在上述步骤2获得的化合物(0.289g),通过与实施例89步骤5相同的方法,得到标题化合物(0.131g)。
1H-NMR(CDCl3)δ:2.15-2.25(1H,m),2.41(3H,d,J=1.2Hz),2.43-2.53(1H,m),2.76-2.83(2H,m),2.92-2.99(2H,m),6.46(1H,d,J=15.7Hz),6.95(1H,d,J=1.2Hz),7.35-7.39(3H,m),7.59(1H,d,J=8.0Hz),8.41(1H,d,J=8.0Hz),8.48(1H,d,J=15.7Hz)。
[实施例94](2E)-[1-{[5-(1-丙烯酰基哌啶-4-基)-3-(2,4,6-三氯苯基)-1,2-唑-4-基]羰基}-3,4-二氢苯并[cd]吲哚-5(1H)-亚基]乙酸
式82
[步骤1]5-{1-[(2-硝基苯基)磺酰基]哌啶-4-基}-3-(2,4,6-三氯苯基)-1,2-唑-4-甲酸五氟苯基酯
在冰冷却下,向参考实施例X-38获得的化合物(20.0g)的二氯甲烷(200ml)溶液中加入N,N-二(丙烷-2-基)乙基胺(9.1ml)和三氟乙酸五氟苯基酯(8.0ml),于室温下将混合物搅拌过夜。将反应液减压浓缩,将获得的残留物经硅胶柱色谱(正己烷/乙酸乙酯)进行纯化,得到标题化合物(24.7g)。
1H-NMR(CDCl3)δ:2.07-2.24(4H,m),2.93-3.08(2H,m),3.61-3.73(1H,m),4.00-4.08(2H,m),7.44(2H,s),7.68-7.63(1H,m),7.69-7.76(2H,m),8.02-8.06(1H,m)。
[步骤2](2E)-[1-{[5-{1-[(2-硝基苯基)磺酰基]哌啶-4-基}-3-(2,4,6-三氯苯基)-1,2-唑-4-基]羰基}-3,4-二氢苯并[cd]吲哚-5(1H)-亚基]乙酸叔丁基酯
在冰冷却下,向参考实施例E-17获得的化合物(5.22g)的N,N-二甲基甲酰胺(97ml)溶液中缓慢加入将氢化钠(55%的油分散液,846mg),于室温下将混合物搅拌30分钟。在冰冷却下,向反应液加入上述步骤1获得的化合物(14.1g),于室温下将混合物搅拌10分钟。在冰冷却下,将反应液用乙酸乙酯稀释,向其中加入冰水。分离两层,将有机层用水和饱和盐水依次洗涤,经无水硫酸钠干燥。减压蒸除溶剂,将获得的残留物经硅胶柱色谱(正己烷/乙酸乙酯)进行纯化,得到标题化合物(11.5g)。
1H-NMR(CDCl3)δ:1.53(9H,s),2.75(2H,t,J=6.7Hz),2.86-2.97(4H,m),2.88-2.95(2H,m),3.20-3.35(3H,m),3.94-4.02(2H,m),6.41(1H,s),6.78(1H,s),7.30-7.36(3H,m),7.46(1H,d,J=7.9Hz),7.60-7.65(1H,m),7.66-7.76(2H,m),8.00(1H,dd,J=7.6,2.1Hz),8.06(1H,d,J=8.5Hz)。
[步骤3](2E)-[1-{[5-(哌啶-4-基)-3-(2,4,6-三氯苯基)-1,2-唑-4-基]羰基}-3,4-二氢苯并[cd]吲哚-5(1H)-亚基]乙酸叔丁基酯
在冰冷却下,向上述步骤2获得的化合物(14.2g)的乙腈(175ml)溶液中加入4-溴苯硫醇(6.61g)和碳酸钾(9.67g),于室温下将混合物搅拌7小时。在冰冷却下,将反应液用乙酸乙酯稀释,向其中加入冰水。分离两层,将有机层用水和饱和盐水依次洗涤,经无水硫酸钠干燥。减压蒸除溶剂,将获得的残留物经硅胶柱色谱(二氯甲烷/甲醇)进行纯化,得到标题化合物(8.66g)。
1H-NMR(CDCl3)δ:1.54(9H,s),1.91-2.17(4H,m),2.71(2H,td,J=11.8,3.2Hz),2.78(2H,t,J=6.7Hz),3.18-3.31(3H,m),3.34(2H,t,J=6.7Hz),6.42(1H,s),6.84(1H,s),7.31-7.37(3H,m),7.47(1H,d,J=7.3Hz),8.10(1H,d,J=7.9Hz)。
[步骤4](2E)-[1-{[5-(1-丙烯酰基哌啶-4-基)-3-(2,4,6-三氯苯基)-1,2-唑-4-基]羰基}-3,4-二氢苯并[cd]吲哚-5(1H)-亚基]乙酸叔丁基酯
在冰冷却下,向上述步骤3获得的化合物(6.13g)的二氯甲烷(98ml)溶液中加入三乙基胺(2.7ml)和丙烯酰氯(0.95ml),将混合物在0℃搅拌20分钟。向反应液中加入冰水,用氯仿萃取。将萃取液用饱和盐水洗涤,经无水硫酸钠干燥。减压蒸除溶剂,将获得的残留物经硅胶柱色谱(正己烷/乙酸乙酯)进行纯化,得到标题化合物(5.93g)。
1H-NMR(CDCl3)δ:1.54(9H,s),2.00(2H,ddd,J=24.6,11.9,3.8Hz),2.08-2.16(2H,m),2.72-2.84(3H,m),3.12-3.25(1H,m),3.34(2H,t,J=6.7Hz),3.42(1H,tt,J=11.5,4.2Hz),4.07-4.18(1H,m),4.70-4.81(1H,m),5.71(1H,dd,J=10.9,1.8Hz),6.30(1H,dd,J=16.3,1.8Hz),6.42(1H,s),6.59(1H,dd,J=16.9,10.3Hz),6.81(1H,s),7.32(2H,s),7.35(1H,t,J=7.9Hz),7.47(1H,d,J=7.3Hz),8.10(1H,d,J=7.9Hz)。
[步骤5](2E)-[1-{[5-(1-丙烯酰基哌啶-4-基)-3-(2,4,6-三氯苯基)-1,2-唑-4-基]羰基}-3,4-二氢苯并[cd]吲哚-5(1H)-亚基]乙酸
使用上述步骤4获得的化合物(6.36g),通过与实施例10的步骤3相同的方法,得到标题化合物(5.28g)。
1H-NMR(CDCl3)δ:2.02(2H,ddd,J=25.1,11.8,3.9Hz),2.09-2.20(2H,m),2.75-2.89(3H,m),3.16-3.28(1H,brm),3.38(2H,t,J=6.3Hz),3.44(1H,tt,J=12.1,3.6Hz),4.07-4.19(1H,m),4.71-4.81(1H,m),5.75(1H,dd,J=10.9,1.8Hz),6.30(1H,dd,J=16.9,1.8Hz),6.54(1H,s),6.59(1H,dd,J=16.6,10.6Hz),6.84(1H,s),7.32(2H,s),7.39(1H,t,J=7.9Hz),7.52(1H,d,J=7.9Hz),8.14(1H,d,J=7.9Hz)。
MS(m/z):624(M+H)+。
使用在参考实施例中获得的化合物,通过与实施例94相同的方法,得到下述化合物。
[表61]
[表62]
[表63]
[表64]
[表65]
[表66]
[表67]
[表68]
[表69]
[表70]
[表71]
[表72]
[表73]
[表74]
[表75]
[实施例132](2E)-3-(1-{[3-(1-丙烯酰基哌啶-4-基)-5-(2,4,6-三氯苯基)-1,2-唑-4-基]羰基}-1H-吲哚-4-基)丙-2-烯酸
式83
[步骤1]4-[4-({4-[(1E)-3-叔丁氧基-3-氧代丙-1-烯-1-基]-1H-吲哚-1-基}羰基)-5-(2,4,6-三氯苯基)-1,2-唑-3-基]哌啶-1-甲酸叔丁基酯
在冰冷却下,向参考实施例X-76获得的化合物(160mg)的二氯甲烷(2ml)溶液中滴加1-氯-N,N,2-三甲基-1-丙烯基胺(0.054ml),于室温下将混合物搅拌1小时。在冰冷却下再向反应液加入1-氯-N,N,2-三甲基-1-丙烯基胺(0.054ml),于室温下将混合物搅拌45分钟。将反应液加至参考实施例E-1获得的化合物(0.082mg)、N,N-二(丙烷-2-基)乙基胺(0.16ml)和4-二甲基氨基吡啶(4mg)的二氯甲烷(1ml)溶液中,于室温下将混合物搅拌过夜。将反应液用乙酸乙酯稀释,用水和饱和盐水依次洗涤,经无水硫酸钠干燥。减压蒸除溶剂,将获得的残留物经硅胶柱色谱(正己烷/乙酸乙酯)进行纯化,得到标题化合物(93mg)。
1H-NMR(CDCl3)δ:1.46(9H,s),1.55(9H,s),1.80-1.93(2H,m),2.01-2.10(2H,m),2.77-2.91(2H,m),3.15-3.25(1H,m),4.07-4.24(2H,m),6.46(1H,d,J=15.7Hz),6.71(1H,d,J=4.2Hz),7.14(1H,d,J=4.2Hz),7.31-7.45(3H,m),7.54(1H,d,J=7.9Hz),7.85(1H,d,J=15.7Hz),8.37(1H,d,J=8.5Hz)。
[步骤2](2E)-3-(1-{[3-(1-丙烯酰基哌啶-4-基)-5-(2,4,6-三氯苯基)-1,2-唑-4-基]羰基}-1H-吲哚-4-基)丙-2-烯酸
于室温下,向上述步骤1获得的化合物(93mg)的二氯甲烷(1ml)溶液中加入三氟乙酸(2ml),于室温下将混合物搅拌1小时。将反应液减压浓缩,在冰冷却下向获得的残留物中加入二氯甲烷(2ml)和饱和碳酸氢钠水溶液(1ml),在剧烈搅拌下加入丙烯酰氯(0.01ml)。于室温下将混合物搅拌10分钟,再加入丙烯酰氯(0.01ml),将混合物搅拌10分钟。将反应液在冰上冷却,然后用1N盐酸使其呈酸性,用二氯甲烷进行萃取。将萃取液用水和饱和盐水依次洗涤,经无水硫酸钠干燥。减压蒸除溶剂,将获得的残留物经硅胶柱色谱(二氯甲烷/甲醇)和制备性薄层色谱(二氯甲烷/甲醇)进行纯化,得到标题化合物(26mg)。
1H-NMR(CDCl3)δ:1.82-2.06(2H,m),2.09-2.26(2H,m),2.80-2.93(1H,m),3.16-3.30(1H,m),3.29-3.40(1H,m),4.03-4.17(1H,m),4.62-4.74(1H,m),5.70(1H,dd,J=10.3,1.8Hz),6.28(1H,dd,J=16.9,1.8Hz),6.55(1H,d,J=15.7Hz),6.59(1H,dd,J=16.9,10.3Hz),6.73(1H,d,J=3.6Hz),7.17(1H,d,J=3.6Hz),7.34(2H,s),7.42(1H,t,J=7.9Hz),7.59(1H,d,J=7.3Hz),8.04(1H,d,J=16.3Hz),8.41(1H,d,J=8.5Hz)。
MS(m/z):598(M+H)+。
[实施例133](2E)-3-(1-{[3-(2,6-二氯苯基)-5-(二甲基氨基)-1,2-唑-4-基]羰基}-1H-吲哚-4-基)丙-2-烯酸
式84
[步骤1](1Z)-2,6-二氯-N-羟基亚氨酰氯
将2,6-二氯苯甲醛肟(5.0g)溶于N,N-二甲基甲酰胺(75ml),在氮环境下,于室温向该溶液中加入N-氯琥珀酸酰亚胺(3.69g)。将混合物搅拌5小时,然后加入水,用乙醚萃取。将萃取液用饱和盐水洗涤,经无水硫酸钠干燥。减压蒸发溶剂后得到标题化合物(6.13g)。
1H-NMR(CDCl3)δ:7.30-7.41(3H,m),9.03(1H,brs)。
[步骤2]5-氨基-3-(2,6-二氯苯基)-1,2-唑-4-甲酸甲基酯
在氮环境中、冰冷却下,向氰基乙酸乙酯(0.25ml)的四氢呋喃(3ml)溶液中滴加甲醇钠(28%甲醇溶液、0.46ml)的甲醇(6ml)溶液,于室温下将混合物搅拌1小时。在冰冷却下,向反应液中滴加上述步骤1获得的化合物(0.50g)的四氢呋喃(1ml)溶液,于室温下将混合物搅拌过夜。减压浓缩反应液,获得的残留物用乙醚稀释,用水和饱和盐水依次洗涤,经无水硫酸钠干燥。减压蒸除溶剂,将获得的残留物经硅胶柱色谱(正己烷/乙酸乙酯)进行纯化,得到标题化合物(0.40g)。
1H-NMR(CDCl3)δ:3.62(3H,s),6.08(2H,brs),7.30-7.41(3H,m)。
[步骤3]3-(2,6-二氯苯基)-5-(二甲基氨基)-1,2-唑-4-甲酸甲基酯
在冰冷却下,向上述步骤2获得的化合物(0.10g)的N,N-二甲基甲酰胺(2ml)溶液中加入碳酸钾(0.24g)和甲基碘(0.49g),将混合物在50℃进行3.5小时搅拌。冷却后,向反应液加入水,用乙酸乙酯进行萃取。将萃取液用饱和盐水洗涤,经无水硫酸钠干燥。减压蒸除溶剂,将获得的残留物经硅胶柱色谱(正己烷/乙酸乙酯)进行纯化,得到标题化合物(0.085g)。
1H-NMR(CDCl3)δ:3.30(6H,s),3.47(3H,s),7.27-7.39(3H,m)。
[步骤4]3-(2,6-二氯苯基)-5-(二甲基氨基)-1,2-唑-4-甲酸
于室温下向上述步骤3获得的化合物(0.51g)的甲醇(10ml)溶液中加入1N氢氧化钠水溶液(10ml),将混合物在50℃搅拌8小时。冷却后,将反应液减压浓缩,向获得的残留物中加入乙酸乙酯,分离两层。在冰冷却下,向水层中加入1N盐酸并中和,用乙酸乙酯进行萃取。将萃取液用饱和盐水洗涤,经无水硫酸钠干燥。经减压蒸除溶剂后得到标题化合物(0.36g)。
1H-NMR(CDCl3)δ:3.29(6H,s),7.26-7.37(3H,m)。
[步骤5](2E)-3-(1-{[3-(2,6-二氯苯基)-5-(二甲基氨基)-1,2-唑-4-基]羰基}-1H-吲哚-4-基)丙-2-烯酸叔丁基酯
在氮环境下,向上述步骤4获得的化合物(0.10g)的苯(3ml)溶液中加入亚硫酰氯(0.12ml)和N,N-二甲基甲酰胺(0.01ml),将混合物加热至回流3小时。冷却后,将反应液减压浓缩,将获得的残留物溶于N,N-二甲基甲酰胺(3ml)。
在氮环境中。冰冷却下,向参考实施例E-1获得的化合物(0.08g)的N,N-二甲基甲酰胺(3ml)溶液中加入氢化钠(55%的油分散液,0.014g),于室温下将混合物搅拌2小时。在冰冷却下,向反应液中加入上述酰氯溶液,于室温下将混合物搅拌16小时。向反应液中加入水,用乙酸乙酯进行萃取。将萃取液用饱和盐水洗涤,经无水硫酸钠干燥。减压蒸除溶剂,将获得的残留物经硅胶柱色谱(正己烷/乙酸乙酯)进行纯化,得到标题化合物(0.165g)。
1H-NMR(CDCl3)δ:1.55(9H,s),3.16(6H,s),6.42(1H,d,J=15.7Hz),6.56(1H,d,J=4.2Hz),7.08-7.21(3H,m),7.30(1H,d,J=7.9Hz),7.34(1H,d,J=3.6Hz),7.45(1H,d=7.3Hz),7.84(1H,d,J=16.3Hz),8.21(1H,d,J=8.5Hz)。
[步骤6](2E)-3-(1-{[3-(2,6-二氯苯基)-5-(二甲基氨基)-1,2-唑-4-基]羰基}-1H-吲哚-4-基)丙-2-烯酸
在冰冷却下,向上述步骤5获得的化合物(0.16g)的二氯甲烷(7ml)溶液中加入三氟乙酸(1ml),于室温下将混合物搅拌18小时。将反应液用二氯甲烷稀释,用水洗涤,经无水硫酸钠干燥。减压蒸除溶剂,将获得的残留物经硅胶柱色谱(二氯甲烷/甲醇)进行纯化,得到标题化合物(0.083g)。
1H-NMR(DMSO-d6)δ:3.04(6H,s),6.58(1H,d,J=15.7Hz),6.81(1H,d,J=3.6Hz),7.30-7.44(4H,m),7.54(1H,d,J=3.6Hz),7.64(1H,d,J=7.9Hz),7.83(1H,d,J=15.7Hz),8.09(1H,d,J=8.5Hz),12.46(1H,brs)。
将实施例133步骤2获得的化合物作为原料,通过与实施例133相同的方法,得到下述化合物。
[表76]
[实施例137](2E)-3-(1-{[3-(2,6-二氯苯基)-5-(顺式-2,6-二甲基哌啶-1-基)-1,2-唑-4-基]羰基}-1H-吲哚-4-基)丙-2-烯酸
式85
[步骤1]3-溴丙-2-炔酸叔丁基酯
向2-丙炔酸叔丁基酯(50.0g)的丙酮(500ml)溶液中加入硝酸银(6.73g),于室温下将混合物搅拌1小时。在水浴中冷却反应液,同时向其中加入N-溴琥珀酸酰亚胺(79.2g),于室温下将混合物搅拌18小时。将反应液经硅藻土过滤,将滤液减压浓缩。向获得的残留物中加入乙醚/正戊烷(1:4)的混合溶剂,将析出的不溶物通过硅藻土过滤。将滤液在减压下浓缩,得到标题化合物(81.3g),无须纯化下使用于下一反应。
[步骤2]5-溴-3-(2,6-二氯苯基)-1,2-唑-4-甲酸叔丁基酯
将上述步骤1获得的化合物(81.3g)和实施例4的步骤1获得的化合物(44.5g)溶于乙酸乙酯(480ml)中,向该溶液中加入水(60ml)并搅拌。在冰冷却下向其中加入碳酸氢钠(50.0g),于室温下将混合物搅拌17小时。将反应液通过硅藻土过滤,将滤液减压浓缩。将获得的残留物溶于乙酸乙酯,用水和饱和盐水依次洗涤,经无水硫酸钠干燥。减压蒸除溶剂,向获得的残留物中加入乙酸乙酯/正己烷,过滤收集产生的固体,得到标题化合物(34.7g)。再将滤液减压浓缩,向获得的残留物中加入正己烷,过滤收集生成的固体,进一步得到标题化合物(15.7g)。
1H-NMR(CDCl3)δ:1.27(9H,s),7.34-7.44(3H,m)。
[步骤3]5-溴-3-(2,6-二氯苯基)-1,2-唑-4-甲酸
在冰冷却下,向上述步骤2获得的化合物(15.7g)的二氯甲烷(50ml)溶液中加入三氟乙酸(10ml),于室温下将混合物搅拌24小时。将反应液用二氯甲烷稀释,用水洗涤,经无水硫酸钠干燥。减压蒸除溶剂,向获得的残留物中加入正己烷,过滤收集生成的固体,得到标题化合物(9.95g)。
1H-NMR(CDCl3)δ:7.36-7.45(3H,m)。
[步骤4](2E)-3-[1-[5-溴-3-(2,6-二氯苯基)-1,2-唑-4-基]羰基}-1H-吲哚-4-基)丙-2-烯酸叔丁基酯
向上述步骤3获得的化合物(9.0g)的二氯甲烷(180ml)悬浮液中加入N,N-二甲基甲酰胺(0.1ml),搅拌混合物。在冰冷却下加入草酰氯(2.5ml),将混合物在30℃搅拌3小时。
在冰冷却下,向参考实施例E-1获得的化合物(6.5g)的N,N-二甲基甲酰胺(180ml)悬浮液中加入氢化钠(55%的油分散液,1.3g),于室温下将混合物搅拌2小时。将反应液在冰上冷却后,使用套管在冰冷却下经30分钟滴加上述酰氯溶液。将混合物升温至室温,同时搅拌17小时,向其中加入水,用乙酸乙酯进行萃取。将萃取液用饱和盐水洗涤,经无水硫酸钠干燥。减压蒸除溶剂,将获得的残留物经硅胶柱色谱(正己烷/乙酸乙酯)进行纯化,得到标题化合物(6.14g)。
1H-NMR(CDCl3)δ:1.57(9H,s),6.48(1H,d,J=16.3Hz),6.87-6.88(1H,m),7.33-7.39(5H,m),7.55(1H,d,J=7.3Hz),7.91(1H,d,J=16.3Hz),8.33(1H,d,J=8.5Hz)。
[步骤5](2E)-3-(1-{[3-(2,6-二氯苯基)-5-(顺式-2,6-二甲基哌啶-1-基)-1,2-唑-4-基]羰基}-1H-吲哚-4-基)丙-2-烯酸叔丁基酯
向上述步骤4获得的化合物(50mg)的N,N-二甲基甲酰胺(1ml)的溶液中加入顺式-2,6-二甲基哌啶(0.024ml),将混合物在80℃搅拌7小时。将反应液减压浓缩,将获得的残留物经硅胶柱色谱(正己烷/乙酸乙酯)进行纯化,得到标题化合物(16.1mg)。
1H-NMR(CDCl3)δ:1.36(6H,d,J=7.3Hz),1.56-1.60(12H,m),1.75-1.90(3H,m),4.27-4.31(2H,m),6.42(1H,d,J=16.3Hz),6.57(1H,d,J=3.6Hz),7.04-7.14(3H,m),7.29(1H,d,J=7.9Hz),7.37(1H,d,J=3.6Hz),7.44(1H,d,J=7.9Hz),7.84(1H,d,J=16.3Hz),8.19(1H,d,J=8.5Hz)。
MS(m/z):594(M+H)+。
[步骤6](2E)-3-(1-{[3-(2,6-二氯苯基)-5-(顺式-2,6-二甲基哌啶-1-基)-1,2-唑-4-基]羰基}-1H-吲哚-4-基)丙-2-烯酸
于冰冷却下,向上述步骤5获得的化合物(16.1mg)的二氯甲烷(2ml)溶液中加入三氟乙酸(0.3ml),于室温下将混合物搅拌过夜。将反应液用二氯甲烷稀释,用水洗涤后,经无水硫酸钠干燥。减压蒸除溶剂,将获得的残留物物制备性薄层色谱(氯仿/甲醇)进行纯化,得到标题化合物(12.2mg)。
1H-NMR(DMSO-d6)δ:1.27(6H,d,J=6.7Hz),1.42-1.86(6H,m),4.05-4.13(2H,m),6.58(1H,d,J=16.3Hz),6.88(1H,d,J=3.6Hz),7.30-7.44(4H,m),7.57(1H,d,J=4.2Hz),7.64(1H,d,J=7.3Hz),7.83(1H,d,J=16.3Hz),8.08(1H,d,J=8.5Hz)。
MS(m/z):538(M+H)+。
通过与实施例137相同的方法,得到下述化合物。
[表77]
[实施例139](2E)-3-[1-({5-[(3R)-3-(氨基甲基)吡咯烷-1-基]-3-(2,6-二氯苯基)-1,2-唑-4-基}羰基)-1H-吲哚-4-基]丙-2-烯酸
式86
[步骤1](2E)-3-[1-({5-[(3R)-3-{[(叔丁氧基羰基)氨基]甲基}吡咯烷-1-基]-3-(2,6-二氯苯基)-1,2-唑-4-基}羰基)-1H-吲哚-4-基]丙-2-烯酸叔丁基酯
向实施例137的步骤4获得的化合物(50mg)和(S)-3-N-丁氧基羰基氨基甲基吡咯烷盐酸盐(35.6mg)的N,N-二甲基甲酰胺(1ml)溶液中加入三乙基胺(0.037ml),于室温下将混合物搅拌过夜。将反应液减压浓缩,将获得的残留物经硅胶柱色谱(氯仿/甲醇)进行纯化,得到标题化合物(60.5mg)。
1H-NMR(CDCl3)δ:1.42(9H,s),1.55(9H,s),1.74-1.87(1H,m),2.10-2.21(1H,m),2.51-2.64(1H,m),3.12-3.32(2H,m),3.34-3.81(3H,m),4.62-4.72(1H,m),6.42(1H,d,J=15.7Hz),6.51(1H,d,J=3.6Hz),7.06-7.21(3H,m),7.28-7.33(2H,m),7.44(1H,d,J=7.3Hz),7.83(1H,d,J=15.7Hz),8.20(1H,d,J=8.5Hz)。
MS(m/z):681(M+H)+。
[步骤2](2E)-3-[1-({5-[(3R)-3-(氨基甲基)吡咯烷-1-基]-3-(2,6-二氯苯基)-1,2-唑-4-基}羰基)-1H-吲哚-4-基]丙-2-烯酸
于冰冷却下,向上述步骤1获得的化合物(60.5mg)的二氯甲烷(3ml)溶液中加入三氟乙酸(0.5ml),于室温下将混合物搅拌8小时。将反应液减压浓缩后,用二氯甲烷稀释,在冰冷却下加入三乙基胺,使其呈弱碱性。减压浓缩,获得的残留物以制备性薄层色谱(氯仿/甲醇/水)进行纯化,得到标题化合物(15.5mg)。
1H-NMR(DMSO-d6)δ:1.20-1.31(2H,m),1.73-1.85(1H,m),2.04-2.16(1H,m),2.63-2.72(3H,m),2.83-2.91(2H,m),3.39-3.76(3H,m),6.56(1H,d,J=15.7z),6.73(1H,d,J=3.6z),7.30-7.35(3H,m),7.45(2H,d,J=3.6z),7.62(1H,d,J=7.3z),7.79(1H,d,J=16.3z),8.09(1H,d,J=8.5z)。
MS(m/z525(M+H)+。
通过与实施例139相同的方法,得到下述化合物。
[表78]
[实施例141](2E)-3-[1-({5-[(2R)-4-丙烯酰基-2-甲基哌嗪-1-基]-3-(2,6-二氯苯基)-1,2-唑-4-基}羰基)-1H-吲哚-4-基]丙-2-烯酸
式87
[步骤1](3R)-4-[4-({4-[(1E)-3-叔丁氧基-3-氧代丙-1-烯-1-基]-1H-吲哚-1-基}羰基]-3-(2,6-二氯苯基)-1,2-唑-5-基]-3-甲基哌嗪-1-甲酸叔丁基酯
向实施例137的步骤4获得的化合物(150mg)的N,N-二甲基甲酰胺(3ml)溶液中加入(3R)-1-叔丁氧基羰基-3-甲基哌嗪(107mg),于室温下将混合物搅拌过夜。将反应液减压浓缩,将获得的残留物经硅胶柱色谱(正己烷/乙酸乙酯)进行纯化,得到标题化合物(137mg)。
1H-NMR(CDCl3)δ:1.32-1.37(3H,m),1.45-1.48(9H,m),1.54-1.57(9H,m),2.95-3.31(2H,m),3.37-3.66(2H,m),3.85-4.35(3H,m),6.42-6.46(1H,m),6.59(1H,s),7.10-7.22(3H,m),7.28-7.35(2H,m),7.45-7.49(1H,m),7.84(1H,d,J=18.7Hz),8.20(1H,d,J=8.5Hz)。
MS(m/z):681(M+H)+。
[步骤2](2E)-3-[1-({3-(2,6-二氯苯基)-5-[(2R)-2-甲基哌嗪-1-基]-1,2-唑-4-基}羰基)-1H-吲哚-4-基]丙-2-烯酸
在冰冷却下,向上述步骤1获得的化合物(137mg)的二氯甲烷(5ml)溶液中加入三氟乙酸(1ml),于室温下将混合物搅拌过夜。将反应液减压浓缩,用二氯甲烷稀释,在冰冷却下加入三乙基胺使其呈弱碱性。减压浓缩反应液,得到标题化合物,其无需纯化下可使用于下一反应。
[步骤3](2E)-3-[1-({5-[(2R)-4-丙烯酰基-2-甲基哌嗪-1-基]-3-(2,6-二氯苯基)-1,2-唑-4-基}羰基)-1H-吲哚-4-基]丙-2-烯酸
将上述步骤2获得的化合物悬浮于二氯甲烷(16ml)中,在冰冷却下向该悬浮液中加入饱和碳酸氢钠水溶液(8ml)和丙烯酰氯(0.034ml),于室温下将混合物搅拌5小时。在冰冷却下再向该反应液中加入丙烯酰氯(0.034ml),于室温下将混合物搅拌过夜。在冰冷却下向反应液加入1N盐酸使其呈酸性,用二氯甲烷萃取。将萃取液经无水硫酸钠干燥。减压蒸除溶剂,将获得的残留物经硅胶柱色谱(二氯甲烷/甲醇)进行纯化,得到标题化合物(86.4mg)。
1H-NMR(DMSO-d6)δ:1.19(3H,d,J=6.7Hz),2.87-3.11(1H,m),3.46-3.61(2H,m),3.94-4.30(4H,m),5.70(1H,d,J=10.9Hz),6.11-6.21(1H,m),6.59(1H,d,J=16.3Hz),6.71-6.83(1H,m),6.86(1H,s),7.31-7.47(4H,m),7.59(1H,s),7.66(1H,d,J=7.9Hz),7.84(1H,d,J=16.3Hz),8.10(1H,d,J=8.5Hz),12.48(1H,s)。
MS(m/z):579(M+H)+。
通过与实施例141相同的方法,得到下述化合物。
[表79]
[表80]
[表81]
[表82]
[表83]
[表84]
[表85]
[表86]
[实施例164](2E)-3-(3-[5-叔丁基-3-(2,4,6-三氯苯基)-1,2-唑-4-基]羰基}-1-甲基-1H-吲哚-7-基)丙-2-烯酸
式88
[步骤1](7-溴-1H-吲哚-3-基)[5-叔丁基-3-(2,4,6-三氯苯基)-1,2-唑-4-基]甲酮
向参考实施例X-5获得的化合物(1.00g)和N,N-二甲基甲酰胺(10μl)的二氯甲烷(20ml)溶液中加入草酰氯(743mg),于室温下将混合物搅拌1小时。将反应液减压浓缩,得到酰氯。
在氮环境中、在冰冷却下,向上述酰氯的二氯甲烷(15ml)溶液中加入氯化铝(781mg),于室温下将混合物搅拌10分钟。
在氮环境下,向7-溴吲哚(1.16g)的二氯甲烷(15ml)溶液中加入上述反应液,于室温下将混合物搅拌40分钟。将反应液倾至冰中,分离两层。将水层用二氯甲烷进行萃取,合并有机层,用饱和盐水洗涤,经无水硫酸钠干燥。减压蒸除溶剂,将获得的残留物经硅胶柱色谱(正己烷/乙酸乙酯)进行纯化,得到标题化合物(514mg)。
1H-NMR(CDCl3)δ:1.43(9H,s),7.18(1H,t,J=7.9Hz),7.29(2H,s),7.45(1H,d,J=7.9Hz),7.66(1H,d,J=3.0Hz),8.29(1H,d,J=7.9Hz),8.67(1H,brs)。
[步骤2](2E)-3-(3-{[5-叔丁基-3-(2,4,6-三氯苯基)-1,2-唑-4-基]羰基}-1H-吲哚-7-基)丙-2-烯酸叔丁基酯
向上述步骤1获得的化合物(510mg)、丙烯酸叔丁基酯(186mg)、N,N-二(丙烷-2-基)乙基胺(0.33ml)和三(2-甲基苯基)膦(88mg)的N,N-二甲基甲酰胺(3.5ml)溶液中加入乙酸钯(33mg),将混合物使用微波装置,在140℃搅拌1小时。冷却至室温后,向反应液中加入乙酸乙酯和饱和盐水,分离两层。将有机层用水和饱和盐水依次洗涤,经无水硫酸钠干燥。减压蒸除溶剂,将获得的残留物经硅胶柱色谱(正己烷/乙酸乙酯)进行纯化,得到标题化合物(430mg)。
1H-NMR(CDCl3)δ:1.44(9H,s),1.55(9H,s),6.42(1H,d,J=16.9Hz),7.28(2H,s),7.31(1H,dd,J=8.2,4.1Hz),7.49(1H,d,J=7.3Hz),7.66(1H,d,J=2.4Hz),7.83(1H,d,J=15.7Hz),8.39(1H,d,J=7.9Hz),8.93(1H,brs)。
MS(m/z):573(M+H)+。
[步骤3](2E)-3-(3-[5-叔丁基-3-(2,4,6-三氯苯基)-1,2-唑-4-基]羰基}-1-甲基-1H-吲哚-7-基)丙-2-烯酸叔丁基酯
在氮环境中、在冰冷却下,向上述步骤2获得的化合物(426mg)的N,N-二甲基甲酰胺(3ml)溶液中加入氢化钠(55%的油分散液,36mg),将混合物搅拌5分钟。向反应液中加入甲基碘(0.14ml),将混合物搅拌1小时。向反应液中加入冰、水和乙酸乙酯,分离两层。将有机层用水和饱和盐水依次洗涤,经无水硫酸钠干燥。减压蒸除溶剂,将获得的残留物经硅胶柱色谱(正己烷/乙酸乙酯)进行纯化,得到标题化合物(354mg)。
1H-NMR(CDCl3)δ:1.45(9H,s),1.54(9H,s),3.99(3H,s),6.32(1H,d,J=15.7Hz),7.25-7.30(3H,m),7.44(2H,d,J=6.7Hz),8.32(1H,d,J=15.7Hz),8.40(1H,d,J=7.9Hz)。
MS(m/z):587(M+H)+。
[步骤4](2E)-3-(3-[5-叔丁基-3-(2,4,6-三氯苯基)-1,2-唑-4-基]羰基}-1-甲基-1H-吲哚-7-基)丙-2-烯酸
向上述步骤3获得的化合物(350mg)的二氯甲烷(5ml)溶液中加入三氟乙酸(1ml),于室温下将混合物搅拌过夜。将反应液倾至水和二氯甲烷的混合液中,分离两层。将有机层用水和饱和盐水依次洗涤,经无水硫酸钠干燥。减压蒸除溶剂,将获得的残留物经硅胶柱色谱(正己烷/乙酸乙酯)进行纯化,得到标题化合物(260mg)。
1H-NMR(CDCl3)δ:1.46(9H,s),4.00(3H,s),6.42(1H,d,J=15.1Hz),7.29(2H,s),7.32(1H,t,J=7.9,3.9Hz),7.45(1H,s),7.49(1H,d,J=7.3Hz),8.45(1H,d,J=7.9Hz),8.52(1H,d,J=15.1Hz)。
MS(m/z):531(M+H)+。
通过与实施例164相同的方法,得到下述化合物。
[表87]
[实施例166](2E)-3-(3-{[5-(1-丙烯酰基哌啶-4-基)-3-(2,4,6-三氯苯基)-1,2-唑-4-基]羰基}-1-甲基-1H-吲哚-7-基)丙-2-烯酸
式89
[步骤1](7-溴-1H-吲哚-3-基)[5-{1-[(2-硝基苯基)磺酰基]哌啶-4-基}-3-(2,4,6-三氯苯基)-1,2-唑-4-基]甲酮
在氮气流中、在冰冷却下,向参考实施例X-38获得的化合物(500mg)的二氯甲烷(5.35ml)悬浮液中加入N,N-二甲基甲酰胺(28μl)和草酰氯(0.153ml),于室温下将混合物搅拌2小时。将反应液减压浓缩,将获得的残留物溶于二氯甲烷(5.35ml)中。在氮气流中、在冰冷却下,向该溶液中加入氯化铝(238mg),于室温下将混合物搅拌15分钟。在冰冷却下向反应液中加入7-溴吲哚(262mg),于室温下将混合物搅拌40分钟。将反应液加于冰中,用二氯甲烷进行萃取,将萃取液经无水硫酸镁干燥。减压蒸除溶剂,将获得的残留物经硅胶柱色谱(正己烷/乙酸乙酯)进行纯化,得到标题化合物(145mg)。
1H-NMR(CDCl3)δ:2.08-2.18(3H,m),2.86-2.93(2H,m),3.25-3.33(1H,m),3.59(1H,dd,J=15.7,9.1Hz),3.97(2H,d,J=12.7Hz),6.90-7.07(1H,m),7.20(1H,q,J=7.9Hz),7.35(2H,dd,J=15.1,8.5Hz),7.45(1H,t,J=6.3Hz),7.53(1H,t,J=3.9Hz),7.59-7.77(2H,m),7.99-8.03(1H,m),8.22(1H,d,J=7.9Hz),8.64(1H,s)。
MS(m/z):739(M+H)+。
[步骤2](2E)-3-(3-{[5-{1-[(2-硝基苯基)磺酰基]哌啶-4-基}-3-(2,4,6-三氯苯基)-1,2-唑-4-基]羰基}-1H-吲哚-7-基)丙-2-烯酸叔丁基酯
使用在上述步骤1获得的化合物(145mg),通过与实施例164的步骤2相同的方法,得到标题化合物(76.7mg)。
1H-NMR(CDCl3)δ:1.59(9H,s),2.08-2.16(4H,m),2.85-2.92(2H,m),3.26-3.35(1H,m),3.97(2H,d,J=13.4Hz),6.41(1H,d,J=15.8Hz),7.30-7.32(2H,m),7.48-7.53(2H,m),7.61-7.63(1H,m),7.69-7.75(2H,m),7.81(1H,d,J=16.4Hz),7.98-8.01(1H,m),8.31(1H,d,J=7.3Hz),8.90(1H,s)。
MS(m/z):787(M+H)+。
[步骤3](2E)-3-(1-甲基-3-{[5-{1-[(2-硝基苯基)磺酰基]哌啶-4-基}-3-(2,4,6-三氯苯基)-1,2-唑-4-基]羰基}-1H-吲哚-7-基)丙-2-烯酸叔丁基酯
使用在上述步骤2获得的化合物(76.7mg),通过与实施例164的步骤3相同的方法,得到标题化合物(44.7mg)。
1H-NMR(CDCl3)δ:1.54(9H,s),2.05-2.12(4H,m),2.87-2.94(2H,m),3.29-3.39(1H,m),3.88-4.04(5H,m),6.32(1H,d,J=15.8Hz),7.29-7.33(3H,m),7.42-7.46(1H,m),7.61-7.72(4H,m),8.00(1H,dd,J=7.3,1.8Hz),8.25-8.35(2H,m)。
MS(m/z):801(M+H)+。
[步骤4](2E)-3-(1-甲基-3-{[5-(哌啶-4-基)-3-(2,4,6-三氯苯基)-1,2-唑-4-基]羰基}-1H-吲哚-7-基)丙-2-烯酸叔丁基酯
使用上述步骤3获得的化合物(44.7mg),通过与实施例94的步骤3相同的方法,得到标题化合物(26.9mg)。
1H-NMR(CDCl3)δ:1.54(9H,s),2.20-2.30(4H,m),2.88-2.97(2H,m),3.11(1H,q,J=7.3Hz),3.42-3.51(3H,m),3.94(3H,s),6.32(1H,d,J=15.2Hz),7.28-7.35(4H,m),7.45(1H,d,J=6.1Hz),8.26-8.36(2H,m)。
MS(m/z):614(M+H)+。
[步骤5](2E)-3-(3-{[5-(1-丙烯酰基哌啶-4-基)-3-(2,4,6-三氯苯基)-1,2-唑-4-基]羰基}-1-甲基-1H-吲哚-7-基)丙-2-烯酸叔丁基酯
使用上述步骤4获得的化合物(26.9mg),通过与实施例94的步骤4相同的方法,得到标题化合物(16.6mg)。
1H-NMR(CDCl3)δ:1.54(9H,s),1.93-2.10(4H,m),2.70-2.90(1H,m),3.11-3.22(1H,m),3.47-3.52(1H,m),3.93(3H,s),4.01-4.17(1H,m),4.74(1H,d,J=10.9Hz),5.71(1H,dd,J=10.6,1.5Hz),6.27-6.35(2H,m),6.59(1H,dd,J=16.6,10.6Hz),7.27-7.35(4H,m),7.42-7.48(1H,m),8.26-8.38(2H,m)。
MS(m/z):670(M+H)+。
[步骤6](2E)-3-(3-{[5-(1-丙烯酰基哌啶-4-基)-3-(2,4,6-三氯苯基)-1,2-唑-4-基]羰基}-1-甲基-1H-吲哚-7-基)丙-2-烯酸
使用上述步骤5获得的化合物(16.6mg),通过与实施例10的步骤3相同的方法,得到标题化合物(10.1mg)。
1H-NMR(DMSO-d6)δ:1.61-1.77(2H,m),2.00(2H,d,J=12.1Hz),2.60-2.74(1H,m),3.01-3.14(1H,m),3.33-3.39(1H,m),3.98-4.11(4H,m),4.33-4.45(1H,m),5.63(1H,dd,J=10.9,2.4Hz),6.06(1H,dd,J=16.6,2.1Hz),6.37(1H,d,J=15.7Hz),6.77(
1H,dd,J=16.6,10.6Hz),7.23(1H,t,J=7.6Hz),7.51(1H,d,J=7.9Hz),7.76-7.80(2H,m),8.01(1H,s),8.12(1H,d,J=7.9Hz),8.33(1H,t,J=16.0Hz),13.5(1H,brs)。
MS(m/z):614(M+H)+。
[实施例167]3-{[5-叔丁基-3-(2,4,6-三氯苯基)-1,2-唑-4-基]羰基}-1-甲基-1H-苯并[g]吲哚-8-甲酸
式90
[步骤1]8-硝基萘-2-羧酸乙基酯
将萘-2-羧酸(10g)的乙酸酐(105ml)溶液冷却至-10℃,经10分钟滴加发烟硝酸(2.71ml)的乙酸酐(13ml)溶液。于室温下将反应液搅拌5小时,然后倾至冰中。过滤除去不溶物,经减压干燥后得到5-和8-硝基萘-2-羧酸的混合物(12.6g)。向获得的固体中加入乙醇(110ml)和浓硫酸(1.2ml),将混合物加热至回流18小时。将反应液减压浓缩,向残留物中加入乙醇,过滤收集产生的固体,粗品产物经硅胶柱色谱(正己烷/乙酸乙酯)进行纯化,得到标题化合物(2.96g)。
1H-NMR(CDCl3)δ:1.46(3H,t,J=7.3Hz),4.47(2H,q,J=7.3Hz),7.66(1H,t,J=7.9Hz),8.02(1H,d,J=9.1Hz),8.16(1H,d,J=8.5Hz),8.22(1H,dd,J=8.8,1.5Hz),8.27(1H,t,J=3.9Hz),9.27(1H,s)。
[步骤2]1H-苯并[g]吲哚-8-甲酸乙基酯
在-45℃,向上述步骤1获得的化合物(1.0g)的四氢呋喃(20ml)溶液中滴加乙烯基氯化镁(14%四氢呋喃溶液、13.7ml),将混合物在与上述相同的温度下搅拌2小时。向反应液加入饱和氯化铵水溶液、饱和盐水和乙酸乙酯,分离两层。将有机层减压浓缩,将获得的残留物经硅胶柱色谱(正己烷/乙酸乙酯)进行纯化,得到标题化合物(342mg)。
1H-NMR(CDCl3)δ:1.44-1.50(3H,m),4.48(2H,q,J=7.3Hz),6.73(1H,t,J=2.7Hz),7.33(1H,t,J=2.7Hz),7.55(1H,d,J=8.5Hz),7.84(1H,d,J=8.5Hz),7.96(1H,d,J=8.5Hz),8.04(1H,dd,J=8.5,1.2Hz),8.86(1H,s),9.22(1H,brs)。
[步骤3]3-{[5-叔丁基-3-(2,4,6-三氯苯基)-1,2-唑-4-基]羰基}-1H-苯并[g]吲哚-8-甲酸乙基酯
向参考实施例X-5获得的化合物(382mg)的甲苯(3.84ml)溶液中加入N,N-二甲基甲酰胺(14μl)和亚硫酰氯(0.4ml),在100℃下将混合物搅拌2小时。将反应液减压浓缩,将获得的残留物溶于二氯甲烷(6.58ml),加入氯化铝(219mg),于室温下将混合物搅拌20分钟。
向上述步骤2获得的化合物(262mg)的二氯甲烷(6.56ml)溶液中加入上述反应液,于室温下将混合物搅拌1.5小时。将反应液倾至冰中,用二氯甲烷萃取,将萃取液经无水硫酸镁干燥。减压蒸除溶剂,将获得的残留物经硅胶柱色谱(正己烷/乙酸乙酯)进行纯化,得到标题化合物(61.1mg)。
1H-NMR(CDCl3)δ:1.41-1.52(12H,m),4.46(2H,q,J=7.1Hz),7.69(1H,d,J=3.0Hz),7.73(1H,d,J=9.1Hz),7.99(1H,d,J=8.5Hz),8.10(1H,d,J=8.5Hz),8.50(1H,d,J=8.5Hz),8.79(
1H,s),9.48(1H,brs)。
[步骤4]3-{[5-叔丁基-3-(2,4,6-三氯苯基)-1,2-唑-4-基]羰基}-1-甲基-1H-苯并[g]吲哚-8-甲酸乙基酯
使用上述步骤3获得的化合物(61.1mg),通过与实施例164的步骤3相同的方法,得到标题化合物(46mg)。
1H-NMR(CDCl3)δ:1.39-1.52(0H,m),4.31(3H,s),4.46(2H,q,J=7.1Hz),7.51(1H,s),7.73(1H,d,J=8.5Hz),8.01(1H,d,J=8.5Hz),8.09(1H,t,J=4.2Hz),8.55(1H,d,J=9.1Hz),9.17(1H,s)。
[步骤5]3-{[5-叔丁基-3-(2,4,6-三氯苯基)-1,2-唑-4-基]羰基}-1-甲基-1H-苯并[g]吲哚-8-甲酸
在冰冷却下,向上述步骤4获得的化合物(46mg)的四氢呋喃(0.48ml)-甲醇(0.48ml)混合液中加入氢氧化锂(13.4mg)的水溶液(0.48ml),将混合物在60℃下搅拌3小时。在冰冷却下向反应液中加入1N盐酸使其呈酸性,将混合物减压浓缩。将获得的残留物用乙酸乙酯萃取,将萃取液经无水硫酸镁干燥。减压蒸除溶剂,将获得的残留物经硅胶柱色谱(二氯甲烷/甲醇)进行纯化,得到标题化合物(37mg)。
1H-NMR(CDCl3)δ:4.33(3H,s),7.27(1H,s),7.53(1H,s),7.75(1H,d,J=8.5Hz),8.06(1H,d,J=8.5Hz),8.15(1H,d,J=8.5Hz),8.59(1H,d,J=8.5Hz),9.26(1H,s)。
[实施例168](2E)-3-(1-{[5-(2-氟丙烷-2-基)-3-(2,4,6-三氯苯基)-1,2-唑-4-基]羰基}-3-甲基-1H-吲哚-4-基)丙-2-烯酸叔丁基胺盐
式91
[步骤1](2E)-3-(1-{[5-(2-氟丙烷-2-基)-3-(2,4,6-三氯苯基)-1,2-唑-4-基]羰基}-3-甲基-1H-吲哚-4-基)丙-2-烯酸叔丁基胺盐
在冰冷却下,向实施例21的步骤2获得的化合物(15g)的2-丙醇(150ml)悬浮液中加入叔丁基胺(2.94ml)的四氢呋喃(15ml)溶液,于室温下将混合物搅拌过夜。过滤悬浮液,将获得的固体在减压下于50℃干燥过夜,得到标题化合物(15.4g)。
1H-NMR(DMSO-d6)δ:1.20(9H,s),1.83(6H,d,J=22.4Hz),2.35(3H,s),6.39(1H,d,J=15.7Hz),7.30(1H,t,J=7.9Hz),7.34(1H,s),7.54(1H,d,J=7.9Hz),7.85(2H,s),7.93(1H,d,J=15.7Hz),8.17(1H,d,J=8.5Hz)。
C25H17Cl3FN2O4·C4H12N分析计算值:C,57.20;H,4.80;Cl,17.46;F,3.12;N,6.90。
实测值:C,57.01;H,4.93;Cl,17.65;F,3.09;N,6.89。
通过与实施例168相同的方法,得到下述化合物。
[表88]
[表89]
[实施例174](2E)-[1-{[5-(1-丙烯酰基哌啶-4-基)-3-(2,4,6-三氯苯基)-1,2-唑-4-基]羰基}-3,4-二氢苯并[cd]吲哚-5(1H)-亚基]乙酸钠盐
式92
[步骤1](2E)-[1-{[5-(1-丙烯酰基哌啶-4-基)-3-(2,4,6-三氯苯基)-1,2-唑-4-基]羰基}-3,4-二氢苯并[cd]吲哚-5(1H)-亚基]乙酸钠盐
向实施例94的步骤5获得的化合物(1.28g)的四氢呋喃(10ml)悬浮液中加入1N乙醇性氢氧化钠溶液(1.8ml),搅拌混合物。确认溶解后,减压除去溶剂、干燥残留物。向其中加入乙醇(10ml),再次减压除去溶剂、干燥残留物。向其中加入乙醇(10ml),在40℃搅拌混合物过夜。加入乙酸乙酯(20ml),于室温下搅拌1小时,过滤收集固体,用乙酸乙酯洗涤。将获得的固体风干过夜,得到标题化合物(1.02g)。
1H-NMR(DMSO-d6)δ:1.63-1.80(2H,m),2.03-2.11(2H,m),2.60-2.64(2H,m),2.71-2.80(1H,m),3.11-3.19(3H,m),3.42-3.52(1H,m),4.09-4.16(1H,m),4.44-4.51(1H,m),5.67(1H,dd,J=10.3,2.4Hz),6.10(1H,dd,J=16.4,2.4Hz),6.38(1H,s),6.82(1H,dd,J=16.4,10.3Hz),7.00(1H,s),7.28(1H,t,J=7.9Hz),7.36(1H,d,J=7.9Hz),7.82(1H,s),7.84(1H,d,J=7.9Hz).
C31H23Cl3N3O5·Na分析计算值:C,57.56;H,3.58;Cl,16.44;N,6.50;Na,3.55.
实测值:C,56.69;H,3.79;Cl,16.13;N,6.28;Na,3.53.
[实施例175](2E)-[1-{[5-(1-丙烯酰基哌啶-4-基)-3-(2,4,6-三氯苯基)-1,2-唑-4-基]羰基}-3,4-二氢苯并[cd]吲哚-5(1H)-亚基]乙酸叔丁基胺盐
式93
[步骤1](2E)-[1-{[5-(1-丙烯酰基哌啶-4-基)-3-(2,4,6-三氯苯基)-1,2-唑-4-基]羰基}-3,4-二氢苯并[cd]吲哚-5(1H)-亚基]乙酸叔丁基胺盐
向实施例94的步骤5获得的化合物(0.64g)中加入叔丁基胺的1mol/L四氢呋喃(1.05ml)溶液,加温使化合物溶解。加入乙酸乙酯(10ml),经遮光后于室温下放置3天。减压浓缩溶剂,干燥残留物,加入乙酸乙酯使固体悬浮。过滤取出固体,用乙酸乙酯洗涤。将获得的固体经风干后,得到标题化合物。
1H-NMR(DMSO-d6)δ:1.21(9H,s),1.68-1.77(2H,m),2.03-2.10(2H,m),2.67(2H,t,J=6.7Hz),2.71-2.79(1H,m),3.11-3.22(3H,m),3.42-3.51(1H,m),4.13(1H,d,J=13.4Hz),4.47(1H,d,J=13.4Hz),5.67(1H,dd,J=10.6,2.1Hz),6.10(1H,d,J=16.4,2.4Hz),6.44(1H,s),6.81(1H,dd,J=17.0,10.3Hz),7.05(1H,s),7.30(1H,t,J=7.9Hz),7.45(1H,d,J=7.3Hz),7.81(2H,s),7.89(1H,d,J=7.9Hz).
C31H23Cl3N3O5·C4H11N的分析计算值:C,60.22;H,5.05;Cl,15.24;N,8.03.
实测值:C,59.61;H,5.05;Cl,15.29;N,7.89.
[试验实施例1]对IDH1R132H和IDH1R132C酶的抑制活性的评价
IDH1R132H蛋白和IDH1R132C蛋白如下制备:将IDH1R132H或IDH1R132C基因整合入pET24b(+)载体(Novagen)中,制备用于6×组氨酸标记的C末端融合的构建物。Rosetta2(DE3)大肠杆菌转化后,用IPTG诱导蛋白的表达。收集大肠杆菌并匀化,随后使用HisTrap HP柱((GE Healthcare Japan Corp.)对6×组氨酸融合蛋白进行亲和纯化,使用Superdex200柱((GE Healthcare Japan Corp.)进行凝胶过滤纯化,获得目标IDH1R132H或IDH1R132C蛋白。
IDH1R132H和IDH1R132C各自可以将2-氧代戊二酸和NADPH转化成D-2-羟基戊二酸(2-HG)和NADP+。因此,通过检测NADPH的水平可以测定IDH1R132H和IDH1R132C酶的活性。
酶的抑制活性评价如下进行:将40μL含有不同浓度的各化合物的各反应液(100mM Tris-HCl(pH 7.5),150mM NaCl,20mM MgCl2,0.5mg/mL牛血清白蛋白,1mM还原型谷胱甘肽,40μM NADPH,0.5mM 2-氧代戊二酸,0.5%二甲基亚砜,50000-0.128nM化合物和作为酶的12nM IDH1R132H或10nM IDH1R132C)加入到384孔板(Greiner Bio One International GmbH,#781096)的各个孔中,将其于室温下温育。在偶尔监测源自NADPH的荧光的同时,在NADPH被消耗完前,通过加入5μL的0.5M EDTA使反应终止。再加入5μL的WST-8试剂(Dojindo Laboratories,#CK04),将其混合。15分钟后,采用读板仪(PerkinElmer,Inc.,EnVision)于450nm测定吸光度。获得的吸光度的值反映了残存的NADPH的量。由吸光度的数据,计算出各浓度的各化合物的酶抑制活性,采用医学统计分析软件GraphPad Prism计算IC50值。
其结果如表90-93所示。
[试验实施例2]采用表达IDH1R132H的TF-1细胞评价2-HG(2-羟基戊二酸)产生的抑制活性
将人红白血病细胞系TF-1(美国典型培养物保藏中心(American Type Culture Collection))在含有10%(最终浓度)胎牛血清(Hyclone,#SH30070.03)和2ng/mL人重组GM-CSF(粒细胞巨噬细胞集落刺激因子)(R&D Systems、#215-GM-050)的RPMI培养基(Life Technologies Corp.,#11875-093)(以下称为生长培养基A)中进行培养。携有IDH1R132H的TF-1细胞系如下制备:将编码人IDH1的IDH1R132H突变体的DNA克隆到逆转录病毒载体pMSCVpuro(Clontech Laboratories,Inc.,#634-401)中。通过脂质转染方法(Invitrogen Corp.,#15338-100)采用构建的载体pMSCVpuro-IDH1R132H转染PT67细胞(Clontech Laboratories,Inc.,#634-401),以便于产生病毒并分泌到上清液中。将含有病毒的培养上清液以2000rpm离心20分钟,获得的离心澄清液作为病毒溶液。将1200000个TF-1细胞悬浮于6mL的生长培养基A中,在10cm板(IWAKI,#3020-100)上铺板。向其中加入最终浓度为4μg/mL的与凝聚胺(Polybrene)(SIGMA ALDRICH Corp.)混合的4mL病毒溶液。病毒感染后,在含有最终浓度为2μg/mL的嘌呤霉素(Invivogen Corp.,#ant-pr)的生长培养基A中选择IDH1R132H表达细胞,随后通过有限稀释法进行克隆以构建表达IDH1R132H的TF-1细胞系TF-1/IDH1R132H。
将在生长培养基A中的40000个细胞/190μL的TF-1/IDH1R132H细胞以190μL/孔的量接种到96孔板(IWAKI、#3860-096)的各孔。然后,向每个孔中加入10μL以生长培养基A稀释成预定浓度的各化合物溶液,将细胞在37℃、5%CO2下培养2天。培养后,将100μL的培养上清液自各个孔转移至96孔圆底板(Corning Inc.,#3799)中。将板以2000rpm离心3分钟,然后,将各个孔中的50μL上清液分注于1.5-m试管(SARSTEDT K.K.,#72.690.001S)。向每个管中加入200μL的乙醇(Wako Pure Chemical Industries,Ltd.,#057-00456),采用旋涡混合器(Pasolina,#NS-8)将试管搅拌30秒,然后于-80℃培养1小时。其后,向每个试管中再加入150μL的超纯水,采用旋涡混合器将试管搅拌30秒,于-20℃保存。将保存的溶液于室温下溶融后,采用内标(3-羟基戊二酸(3-HG))溶液进行稀释,通过固相萃取(Oasis MAXμElution(Waters Corp.))制备成用于浓度测定的样品。将10μL的各个样品注入UPLC(Waters Corp.)。将含有0.5%甲酸的50%甲醇作为移动相(流速:0.6mL/min),待分析物采用保持于40℃的分析柱(Hypercarb(2.0×150mm,粒径5μm,Thermo Fisher Scientific Inc.))分离,然后导入质谱分析装置(Xevo TQ MS(Waters Corp.)。对于通过电喷雾离子化方法源自2-HG离子化的的负离子(质荷比:147)所产生的质荷比为129的产品离子(保留时间:1.0分)进行观测,通过由2-HG标准溶液制备的标准曲线计算溶液中的2-HG浓度。通过化合物浓度对各浓度的相对2-HG浓度(相对于未加入化合物的细胞中的2-HG浓度的%)的单侧对数坐标图计算各个化合物对2-HG产生达到50%抑制的浓度(IC50值)。
该结果如表90~93所示。
[试验实施例3]使用HT1080细胞评价2-HG产生的抑制活性
将表达IDH1的突变体IDH1R132C的人纤维肉瘤细胞系HT-1080(美国典型培养物保藏中心(American Type Culture Collection))在含有最终浓度为10%胎牛血清(Hyclone,#SH30070.03)的RPMI培养基(Life Technologies Corp.,#11875-093)(以下称为生长培养基B)中进行培养。将生长培养基A中的2000个细胞/100μL的HT1080细胞以100μL/孔的量接种于96孔板(IWAKI,#3860-096)。培养1天后,自各孔移出培养上清液。向每个孔中加入100μL的生长培养基B,再次移出生长培养基B。然后,向各孔中加入100μL的生长培养基B,再加入25μL的用生长培养基B稀释至预定浓度的各个化合物的溶液,将细胞在37℃、5%CO2下培养2天。培养后,自各个孔中将90μL的培养上清液移至96孔圆底板(Corning Inc,#3363)。将板于室温下以2000rpm离心3分钟后,自各孔将澄清液50μL分注于96孔深孔培养板(Porvair plc,#219009)。将板采用200μL的甲醇(Wako Pure Chemical Industries,Ltd.,#131-01826)搅拌,移液,然后于-80℃培养20-60分钟。然后,向每个孔中再加入150μL的超纯水,将混合物通过移液搅拌,于-20℃保存。
将保存的溶液于室温下融解。然后,采用与[试验实施例2]相同的方法,计算该溶液中的2-HG浓度。通过化合物浓度对各浓度的相对2-HG浓度(相对于未加入化合物的细胞中的2-HG浓度的%)的单侧对数坐标图计算每个化合物对2-HG产生达到50%抑制的浓度(IC50值)。
该结果如表90~93所示。
[试验实施例4]TF-1/IDH1R132H细胞生长的抑制活性的评估
将培养于生长培养基A的TF-1/IDH1R132H细胞的细胞悬浮液30mL分注于50mL的每个试管(IWAKI,#2345-050)中。将试管于室温下以1200rpm离心5分钟,移出上清液。将细胞悬浮于30mL的生长培养基B中,使细胞悬浮,于室温下以1200rpm离心5分钟,移出上清液。该操作再重复进行2次。将获得的TF-1/IDH1R132H细胞采用生长培养基B调节至2000个细胞/100μL的细胞浓度。
制备含有最终浓度为10%胎牛血清、4.44pg/mL人重组体GM-CSF的RPMI培养基(以下称为增生培养基C)。将90μL的增生培养基C分配到96孔板(COSTAR,#3904)的各孔中。然后,向每个孔中加入10μL的用生长培养基B稀释至预定浓度的各个化合物的溶液。再向每个孔中加入100μL的制备的TF-1/IDH1R132H细胞悬浮液,将其在37℃、5%CO2下培养2星期。培养后,根据随附的手册使用ATPlite 1步发光分析系统(Luminescence Assay System)(PerkinElmer,Inc.,#6016739)使得细胞进行反应。然后,使用读板仪(PerkinElmer,Inc.,EnVision)测定各孔发光量。通过化合物加入组(T)、化合物未加入组(C)和细胞未加入组(B)的发光量,根据下式计算细胞存活率:
细胞存活率%=(T-B)/(C-B)×100
通过化合物的浓度对每个浓度对应的细胞存活率的单侧对数坐标图计算每个化合物对TF-1/IDH1R132H细胞生长达到50%抑制的浓度(IC50值)。。
其结果如表90~93所示。
[试验实施例5]使用HT1080移植瘤小鼠评价2-HG产生抑制活性
将表达IDH1的突变体IDH1R132C的人纤维肉瘤细胞系HT-1080(美国典型培养物保藏中心)在含有最终浓度为10%胎牛血清(Hyclone#SH30070.03)和青霉素-链霉素(Life Technologies Corp.#15070-063)的RPMI培养基(Life Technologies Corp.#11875-093)中进行培养。
将HT1080细胞采用PBS(Life Technologies Corp.,#14190―250)调节至4×107个细胞/mL的细胞浓度,将其以100μL/小鼠的量皮下植入Balb/c裸鼠(Charles River Laboratories Japan,Inc.)的右腋窝部位。成功植入肿瘤的HT1080移植瘤小鼠通过口服管饲化合物75mg/kg,每天2次,合计3天。将化合物采用均质仪以7.5mg/mL的量悬浮于载体溶液中。然后,将该悬浮液在-20℃下保存,于给药前刻立即融解,给药于小鼠。用于试验的阴性对照组通过口服管饲给药。小鼠体重使用小动物用自动天秤进行测定。以10mL/kg体重的剂量给予剂量溶液。
最终给药后6小时,将每只小鼠采用二氧化碳安乐死。收集部分肿瘤,用电子天秤测定其重量。然后,在干冰上将肿瘤冷冻。向冷冻肿瘤中加入肿瘤重量14倍量的甲醇。将获取的肿瘤采用Beads Shocker(Yasui Kikai Corp.)进行均质化,然后在-20℃的冰箱中培养15分钟。采用高速冷冻微型离心机以15000rpm、于4℃下离心15分钟后,向150μL获得的上清液中加入加250μL的甲醇和400μL的超纯水。制备的样品进行2-HG测定。溶液中的2-HG浓度采用与[试验实施例2]的相同方法计算。通过化合物给药组(T)和阴性对照组(C)的2-HG浓度根据下式计算2-HG产生抑制率:
2-HG产生抑制率%=T/C×100
结果如表90~93所示。
[试验实施例6]携有4个基因包括IDH1R132H的AML细胞的构建与2-HG的测定
用于基因转导的病毒如下制备:首先,制备下面所示载体用于逆转录病毒制备。所有使用的基因皆为在急性髓细胞白血病(AML)中常见的高频率突变的突变型基因。pMy-NPMc-ires-EGFP通过将突变型NPM1基因(细胞质核磷蛋白1(cytoplasmic nucleophosmin1);以下称为NPMc)插入pMy-ires-EGFP载体(Cell Biolabs,Inc.,San Diego,CA,USA)的多克隆位点而制备。pGCDN-IDH1/R132H-ires-NGFR通过将IDH1/R132H突变基因插入pGCDN-ires-NGFR载体(由Shimon Sakaguchi教授,Kyoto University提供)的多克隆位点而制备。pMSCV-DNMT3A(DNA甲基转移酶3A)/R882H通过将DNMT3A/R882H突变体基因插入pMSCVpuro载体(Takara Bio Inc.)而制备。pMSCV-FLT3/ITD通过将FLT3/ITD突变基因(FMS样酪氨酸激酶3/内部串联重复(Internal Tandem Duplication))插入pMSCVneo载体(Takara Bio Inc.)而制备。
将PLAT-E细胞(由Toshio Kitamura教授提供,The Institute of Medical Science,The University of Tokyo)通过采用GeneJuice(Merck)的4种逆转录病毒载体(pMy-NPMc-ires-EGFP、pGCDN-IDH1/R132H-ires-NGFR、pMSCV-DNMT3A/R882H和pMSCV-FLT3/ITD)的每一种进行转染。48小时后,回收10ml的每一种含有逆转录病毒的培养上清液。向其中加入3.3ml的PEG浓缩液(30%PEG-8000、0.4M NaCl和40mM HEPES[pH 7.4]),将混合物于4℃静置过夜。将样品离心(1500rpm×45分钟,4℃)得到沉淀。向Stempro-34溶液(Invitrogen Corp.)(以下称为Stempro培养基)中加入50ng/ml SCF、10ng/ml IL-3和10ng/ml OSM。将病毒沉淀悬浮于200μl的Stempro介质中,分配100μl的每一种悬浮液,在液氮中急速冷冻,在-80℃冰箱中保存。
基因转导和首次骨髓移植(primary bone marrow transplantation)如下进行:
自8周龄的NPM+/-小鼠(TaconicArtemis GmbH)的下肢回收骨髓细胞。溶血处理后,使用CD117MACS珠(Miltenyi Biotec K.K.),回收c-Kit+阳性细胞。向用RetroNectin(Takara Bio Inc.)涂布的24孔板的每个孔中加入pMy-NPMc-ires-EGFP的浓缩病毒100μl,向每个孔中加入悬浮于400μl的Stempro介质中的c-Kit+细胞4×105个,在恒温培养器中培养。半天后,使用PBS回收细胞,将细胞悬浮于400μl的Stempr介质中。向用Retronectin(Takara Bio Inc.)包被的24孔板的每个孔中加入pGCDN-IDH1/R132H-ires-NGFR的浓缩病毒100μl,向每个孔中加入细胞400μl,在恒温培养器进行培养。半天后,以与上述同样的方法进行pMSCV-DNMT3A/R882H的病毒感染。半天后,以与上述同样的方法进行pMSCV-FLT3/ITD的病毒感染,从而制备携有4种基因的转染细胞。
将病毒感染的细胞悬浮于600μl的Stempro-34溶液中。将300μl细胞悬浮液经静脉注射到暴露于放射线(950Gy)的2只小鼠的每一只的尾部。每4周采取末梢血,通过FACS分析EGFP与NGFR的表达。EGFP阳性细胞被视为AML细胞。确认AML细胞的移植后,回收骨髓细胞,将其以5×106个细胞/ml悬浮于Bambanker(NIPPON Genetics Co.,Ltd.),储存首次移植小鼠的细胞。
二次骨髓移植和化合物给药如下进行:将每个首次移植小鼠的骨髓细胞以1×105个细胞静脉注射至暴露于放射线(600Gy)的每只小鼠的尾部。二次移植后4周,收集所有小鼠的血液,随后进行外周血细胞的FACS分析。将小鼠随机分组以使得EGFP-阳性的AML细胞的比例不会出现偏差。移植6周后,将实施例168和实施例94的化合物以7.5、15和30mg/mL(以游离形式计算)的浓度方便悬浮于0.5%甲基纤维素中。将每种悬浮液以10mL/kg的剂量给药于小鼠。化合物以每天2次的间隔给药,共计3次。于最终给药的6、16、24小时后,回收血浆和骨髓细胞。向20μL血浆中加入水80μL制备100μL的溶液。将1×106个骨髓细胞悬浮于100μL的PBS中。向100μL的血浆或骨髓的溶液中加入400μL的乙醇,将其混合,随后于-20℃温育1小时。然后,再向其中加入300μL的水,于4℃以15000rpm离心20分钟后,回收上清液并于-20℃保存。
将保存的溶液采用与试验实施例2同样的方法进行处理,测定2-HG。
如图1所示,实施例168和实施例94的化合物的各自给药导致了血浆中2-HG浓度和骨髓细胞中2-HG水平显著降低。
[试验实施例7]对携有4种基因包括IDH1R132H的AML小鼠模型的抗肿瘤活性的测定
含有实施例94化合物的饲料是基于CRF-1(Oriental Yeast Co.,Ltd.)并通过以0.3%(游离形式的重量比)的比例加入化合物制备(Oriental Yeast Co.,Ltd.)。
将在试验实施例6中所建立的首次移植小鼠的骨髓细胞各自以1×105个细胞经静脉注射至暴露于放射线照射(600Gy)的各个小鼠的尾部。移植6周后,回收某些小鼠的骨髓细胞,确认EGFP-阳性AML细胞的比例。80%的骨髓细胞的平均为EGFP阳性。因此,确认移植细胞在骨髓中显著增加。
移植8周后,收集所有小鼠的血液,随后进行外周血细胞的FACS分析。为使EGFP阳性的AML细胞的比例不会偏差,将小鼠随机分组(4只小鼠给予对照饲料,4只小鼠给予补充有实施例94化合物的饲料)。使用含有实施例94化合物的饲料,开始进行混合饲料的给药。
给予混合饲料4周后,收集各小鼠外周血和骨髓细胞并进行FACS分析。检测EGFP信号阳性的AML细胞的比例。
如图2所示,对于4只中3只小鼠而言,含有实施例94化合物的混合饲料经4周给药导致了骨髓中和外周血中EGFP阳性的AML细胞的比例有大幅度减少。
[试验实施例8]对于IDH1/R132H突变型神经胶质母细胞瘤A1074移植小鼠模型中的抗肿瘤活性的测定
将由Peter Collins教授(Department of Pathology,University of Cambridge)提供的IDH1/R132H突变型人神经胶质母细胞瘤的小鼠皮下移植物的传代样本(A1074)根据文献方法(Goike HM:存活的人胶质母细胞瘤移植瘤的冷冻保存(Cryopreservation of viable human glioblastoma xenografts).Neuropathol.Appl.Neurobiol.26:172-176,2000)在37℃水浴下快速溶融,用手术刀切出含有肿瘤边缘和中心部位的5mm的切片。然后,将这些切片皮下移植至NOD/SCID小鼠的腹部。当肿瘤成为1cm大小时,将小鼠通过颈椎脱臼进行安乐死并摘出肿瘤,然后移植到NOG小鼠(Central Institute for Experimental Animals),用于创建A1074小鼠移植模型。
将分离自A1074肿瘤的5mm切片移植至15只NOG小鼠每只的腹侧皮下。使用适宜的光标卡尺测量肿瘤大小,肿瘤体积(mm3)根据(肿瘤长度)×(肿瘤宽度)2/2的计算式计算,用于确认肿瘤的生长和药效。
移植4周后,将小鼠分为3组,每组包括5只小鼠。然后,采用含有实施例168化合物的饲料、含有实施例94化合物的饲料和对照饲料进行4星期的混合饲料给药。含有每种化合物的饲料均是以CRF-1(Oriental Yeast Co.,Ltd.)为基础并通过以0.3%(游离形式的重量比)的比例加入化合物制备(Oriental Yeast Co.,Ltd.)。进行4星期的混合饲料给药后,将每只小鼠通过颈椎脱臼使其安乐死。摘出肿瘤并测定其重量。
该试验中的各组肿瘤体积的平均值和标准差如图3所示。给予混合饲料4周后的肿瘤重量如图4所示。在本实验中,发现实施例168化合物和实施例94化合物能够抑制肿瘤生长。
[工业实用性]
本发明的化合物或其药学上可接受的盐能够抑制可以表达突变型IDH1蛋白的肿瘤的生长,因此可以作为医疗领域中的抗肿瘤药物。同样,本发明的化合物或其药学上可接受的盐也可以作为抑制突变型IDH1蛋白的活性的药物,用于研究的目的。
- 还没有人留言评论。精彩留言会获得点赞!