齐格勒‑纳塔催化剂及其制备的制作方法
本发明涉及用于生产烯烃聚合物的固体齐格勒-纳塔催化剂组分和所述催化剂组分的制备。此外,本发明涉及包含所述固体催化剂组分、作为助催化剂的第13族金属化合物和任选地外部添加剂的齐格勒纳塔催化剂。本发明还涉及所述催化剂组分在生产烯烃聚合物尤其是具有所希望的特性的乙烯聚合物中的用途。
背景技术:
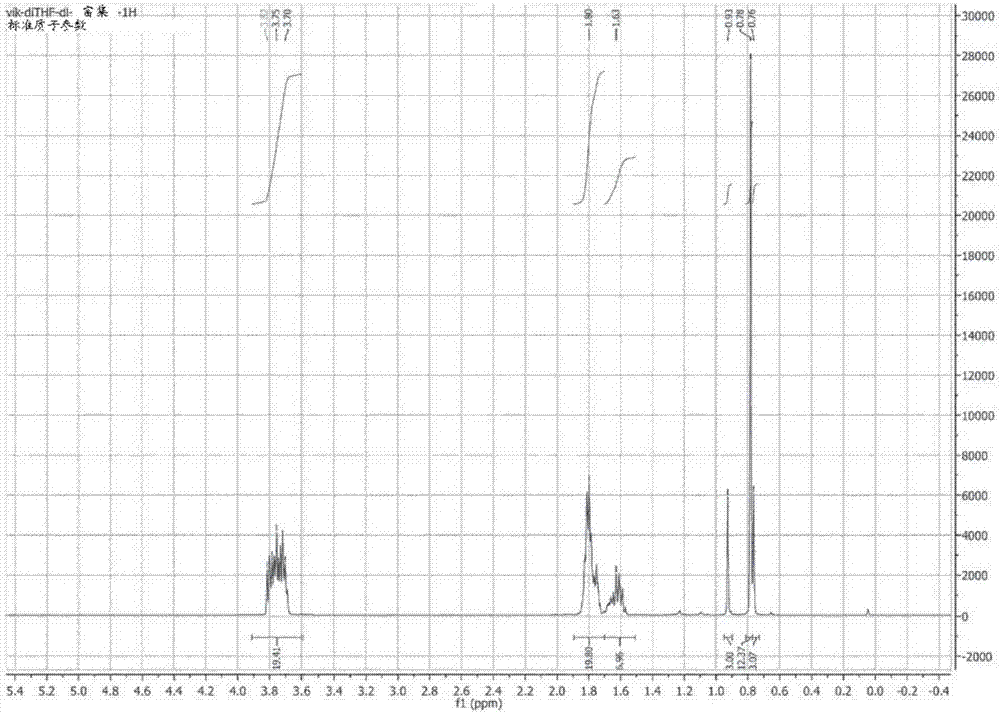
:齐格勒-纳塔(zn)型聚烯烃催化剂在生产烯烃聚合物(如乙烯(共)聚合物)领域中是公知的。通常,这些催化剂至少包含由周期表(iupac,无机化学命名法(nomenclatureofinorganicchemistry),1989)第4至6族的过渡金属化合物、周期表(iupac)第1至3族的金属化合物以及任选地周期表(iupac)第13族的化合物和任选地内部有机化合物如内部电子供体形成的催化剂组分。zn催化剂还可以包含一种或多种另外的催化剂组分,例如助催化剂和任选地外部添加剂。已经开发了种类繁多的齐格勒-纳塔催化剂,以满足反应特性和生产具有所希望的物理和机械性能的聚(α-烯烃)树脂的不同需求。典型的齐格勒-纳塔催化剂含有负载在颗粒支持体上的镁化合物、铝化合物和钛化合物。通常使用的颗粒支持体是无机氧化物类型的支持体,例如二氧化硅、氧化铝、二氧化钛、二氧化硅-氧化铝和二氧化硅-二氧化钛,典型地为二氧化硅。催化剂可以通过使载体与上述化合物顺序接触来制备,例如,如ep688794和wo99/51646中所述的。可替代地,可以通过首先从各组分制备溶液并且然后使该溶液与载体接触来制备,如wo01/55230中所述的。另一组典型的齐格勒-纳塔催化剂基于二卤化镁(典型地为mgcl2),其含有钛化合物和任选地第13族化合物,例如铝化合物。这类催化剂披露在例如ep376936、wo2005/118655和ep810235中。要求上述zn催化剂可用于烯烃聚合,例如生产乙烯(共)聚合物。然而,尽管现有技术的许多催化剂针对许多应用显示出令人满意的特性,但是仍需要修饰和改进催化剂的特性和性能以实现希望的聚合物特性,并且拥有在希望的聚合过程中具有所希望性能的催化剂。关于控制分子量(mw)、聚合物分子量分布(mwd)和共聚单体含量的可能性的氢和共聚单体响应以及因此催化剂灵活性是催化剂性能的一般指标。因此,与这些特性相关的问题表明催化剂的性能特性。此外,公知如果所希望的是高分子量聚合物,并且不能再减少氢的量,则在聚合中可以使用外部添加剂。然而,在这种情况下,通常以牺牲催化剂生产率为代价来生产聚合物。已经尝试通过修饰催化剂来找寻解决方案。修饰催化剂的一种方式是使用内部有机化合物。然而,即使例如聚合物的分子量得到改善,也通常以牺牲一些其他特性(一般为催化剂生产率和共聚单体响应)为代价。内部有机化合物可以是内部电子供体或影响催化剂性能的其他化合物,并且外部添加剂包含例如外部电子供体和/或烷基卤化物。us5,055,535披露了使用包含选自单醚(例如四氢呋喃)的电子供体的zn催化剂来控制聚乙烯均聚物和共聚物的分子量分布(mwd)的方法。在助催化剂存在下将单醚加入到催化组分中,并且进一步的特征是在介质中不存在助催化剂的情况下,在任何情景下,单醚都不会与催化组分接触。wo2007051607a1提出了通过使用烷基醚型内电子供体(优选四氢呋喃)来修改多峰乙烯聚合物的特性以修饰zn催化剂组分从而影响较高分子量(hmw)组分的分子量分布(mwd)的可能性。wo2004055065披露了用来制备乙烯与α-烯烃的共聚物的包含处于特定摩尔比的ti、mg、卤素和电子供体的固体催化剂组分,其中所述α-烯烃沿着聚合物链均匀分布。该电子供体(ed)优选为醚,如四氢呋喃。所述催化剂组分与烷基铝化合物和任选地外部电子供体一起用在聚合中。据说可选的外部电子供体等同于或不同于催化剂组分中使用的ed。ep0376936披露了一种mgcl2负载zn催化剂,其中将喷雾干燥的mgcl2/醇载体材料用ia至iiia族(周期表(iupac,无机化学命名法,1989)第1、2和13族)化合物处理)进行处理,然后用钛化合物钛化,任选地在内电子供体的存在下。与ticl4一起或在加入ticl4后加入任选的内供体化合物(thf或二异丁基邻苯二甲酸酯在使用内电子的那些实例中给出)。然而,ep0376936的供体修饰催化剂的活性远低于没有供体的原始催化剂的活性。此外,在供体处理步骤中,使用10wt%的三乙基铝溶液和多次烃洗涤,导致大量的有机溶剂浪费。wo2014004396a1披露了一种催化剂组分,其中双杂环化合物被用作内部或外部供体。该催化剂被用于丙烯聚合。ep2746306披露了一种负载型齐格勒-纳塔催化剂组分,其包含选自双环醚的内电子供体。ep2746306的催化剂是通过在颗粒支持体上沉积第1至3族金属的可溶性烷氧基化合物、第13族金属化合物、内部电子供体和第4至6族的过渡金属化合物,或者可替代地通过接触可溶性烃氧基镁化合物、电子供体溶液和烷基氯化铝化合物形成沉淀的的负载材料来制备的。在沉淀和合适的洗涤步骤之后,用钛化合物处理所得的固体负载材料以获得催化剂组分。在这种情况下,以催化剂生产率为代价改进了聚合物的分子量。此外,沉淀的mgcl2基催化剂的催化剂性能和形态典型地对制备条件甚至很小的变化敏感,尤其是在大规模生产中。虽然在齐格勒-纳塔催化剂制备上有许多开拓性工作,但仍有改进的余地。如上所述,这些方法中的一些对制备条件特别敏感和/或形成大量的废料,这些在大规模制备催化剂时是缺点。催化剂合成程序的修饰可能不利地影响后续催化剂的生产率,从而不能满足商业规模的生产。此外,催化剂形态可能难以控制,尤其是在大规模生产中。除了对催化剂特性和性能的需求外,商业规模的催化剂制备应尽可能简单和稳健。此外,从健康、安全和环境的角度来看,制备中使用的化学品应被视为是安全的。因此,将希望的是,找到一种更稳健的方法来制备催化剂,其允许生产大规模的催化剂,其对形态变化于催化剂制备条件和化学品的变化敏感性较小。此外,希望可以避免合成期间的大量废料。此外,从商业角度来看,催化剂应显示出可重复的组成和性能。还需要找到能够产生具有更宽的熔体流动速率(mfr)和密度窗口的共聚物的催化剂,使得有可生产具有窄mwd(分子量分布)和与低熔融温度结合的高共聚单体含量的高分子量共聚物。并且最终,催化剂应具有一定水平的生产率,这使得其在商业聚合工艺中是有用的,同时生产宽范围分子量聚合物。基于现有技术的教导,似乎供体修饰可能导致一些特性的改进。然而,这些改进通常以催化剂生产率和共聚单体响应为代价。通过沉淀方法制备的mgcl2基催化剂典型地对制备条件的变化敏感。技术实现要素:现在已经出人意料地发现,当使用用特定的内部有机化合物修饰并通过如下所述的定义方法制备的固体mgcl2基催化剂组分时,可以解决现有技术的问题。因此,本发明的目的是提供制备固体mgcl2基催化剂组分的新方法。本发明还涉及固体mgcl2基催化剂组分和通过本发明方法制备的固体mgcl2基催化剂组分。此外,本发明涉及包含所述固体mgcl2基催化剂组分、助催化剂和任选地外部添加剂的催化剂。本发明的另一个目的是通过本发明的方法制备的固体mgcl2基催化剂组分在烯烃聚合过程中的用途。在本披露中,术语内部有机化合物涵盖但不限于内部电子供体,该短语在专利文献中被广泛使用。所述内部有机化合物表示作为该固体催化剂组分的部分的化合物,即在固体催化剂组分的合成过程中加入。外部添加剂覆盖任何添加剂,覆盖但不限于外部电子供体,并且表示不是该固体催化剂组分的部分,而是作为分离组分进料到聚合过程中的组分。短语载体和负载在本披露中具有相同的含义。具体实施方式因此,本发明涉及一种生产固体mgcl2负载型催化剂组分的方法,该方法包含以下步骤a)提供mgcl2*mroh加合物的固体载体颗粒b)用第13族金属的化合物预处理步骤a)的固体载体颗粒c)用第4至6族的过渡金属化合物处理步骤b)的经预处理的固体载体颗粒d)回收该固体催化剂组分其中在步骤c)中处理这些固体载体颗粒之前,使这些固体载体颗粒与该式(i)的内部有机化合物或其异构体或混合物接触并且其中在式(i)中,r1至r5相同或不同,并且可以是氢、直链或支链c1至c8-烷基或c3-c8-亚烷基,或者r1至r5中的两个或更多个可以形成环,这两个含氧环分别是饱和的或部分不饱和的或不饱和的,并且在该加合物mgcl2*mroh中的r是具有1至12个c原子的直链或支链烷基,并且m是0至6。因此,在使用第4至6族的过渡金属化合物处理固体载体颗粒之前,使该式(i)的内部有机化合物与固体载体颗粒接触。因此,所述内部有机化合物可以在步骤b)之前与固体载体颗粒接触,即在用第13族金属化合物预处理固体载体颗粒之前,或与所述预处理步骤同时,或者在步骤b)之后,但在用第4至6族的过渡金属化合物处理固体载体颗粒之前。本发明还涉及通过上述方法制备的固体催化剂组分。此外,本发明提供了包含如上定义的制备的固体催化剂组分、助催化剂和任选地外部添加剂的齐格勒-纳塔催化剂。此外,本发明的一个目的是根据本发明的催化剂在生产乙烯聚合物的过程中的用途。本发明的催化剂或通过本发明方法生产的催化剂尤其适用于以多阶段工艺生产乙烯共聚物。下面将关于特定的优选实施例更详细地描述本发明。在从属权利要求及以下说明中描述了优选实施例。如此处所使用的,术语齐格勒纳塔(zn)催化剂组分旨在覆盖包含负载于mgcl2基载体上的周期表(iupac,无机化学命名法(nomenclatureofinorganicchemistry),1989)第4至6族过渡金属化合物、第13族金属化合物和内部有机化合物的催化剂组分。二卤化镁用作生产载体的原料。用于本发明的固体载体中是如下的载体,其中醇与二卤化镁配位,优选mgcl2。将mgcl2与醇(roh)混合,并且根据公知的方法形成固体载体mgcl2*mroh。作为实例,喷雾干燥或喷雾结晶方法可用于制备卤化镁。球形和颗粒mgcl2*mroh载体材料适合于在本发明中使用。在生产mgcl2*mroh载体材料中的醇是醇roh,其中r是含有1至12个碳原子,优选1至8个碳原子,如1至4个碳原子的直链或支链烷基。典型地使用乙醇。在mgcl2*mroh中,m为0至6,更优选为1至4,尤其为2,7至3,3。mgcl2*mroh可从商业来源获得,或可以通过现有技术中描述的方法制备。mgcl2*mroh载体的制备方法在几个专利中有描述,例如ep-a-0376936、ep-a-0424049、ep-a-655073、us4,071,674和ep-a-0614467中,这些专利通过引用结合在此。本发明的固体载体颗粒可以由mgcl2*mroh组成。步骤b)中使用的第13族金属化合物优选为铝化合物。特别优选地,该铝化合物是式al(烷基)xx3-x(ii)的铝化合物,其中每个烷基独立地为具有1至12个碳原子,优选1至8个碳原子的烷基,更优选1至6个碳原子,x是卤素,优选氯并且1<x≤3的烷基。该烷基可以是直链的、支链的或环状的,或这类基团的混合物。优选的铝化合物是二烷基氯化铝或三烷基铝化合物,例如二甲基氯化铝、二乙基氯化铝、二异丁基氯化铝和三乙基铝或其混合物。最优选地,该铝化合物是三烷基铝化合物,尤其是三乙基铝化合物。第4至6族的过渡金属化合物优选为第4族过渡金属化合物或钒化合物,并且更优选为钛化合物。特别优选地,该钛化合物是式xyti(or8)4-y的含卤素钛化合物,其中r8是c1-20烷基,优选为c2-10并且更优选为c2-8烷基,x是卤素,优选氯,并且y为1、2、3或4,优选3或4,并且更优选为4。合适的钛化合物包括三烷氧基一氯化钛、二烷氧基二氯化钛、烷氧基三氯化钛和四氯化钛。优选使用四氯化钛。内部有机化合物选自式(i)的双环醚化合物或者其异构体或混合物:其中在式(i)中,r1至r5相同或不同,并且可以是氢、直链或支链c1至c8-烷基或c3-c8-亚烷基,或者r1至r5中的两个或更多个可以形成环,并且其中这两个含氧环分别是饱和的或部分不饱和的或不饱和的,并且加合物mgcl2*mroh中的r是具有1至12个碳原子的直链或支链烷基,并且m是0至6。优选的直链或支链c1至c8-烷基的实例是甲基、乙基、正丙基、异丙基、正丁基、仲丁基、叔丁基、戊基和己基。优选的c3-c8-亚烷基的实例是亚戊基和亚丁基。这两个r1优选相同并且是直链c1至c4-烷基,更优选甲基或乙基;或者两个r1与它们附接的碳原子一起形成具有3至7个碳原子的环,优选环戊基或环己基环。最优选地,两个r1都是甲基。r2至r5相同或不同,并且优选为h或c1至c2-烷基,或者r2至r5残基中的两个或更多个可以形成环。如果一个或多个环由残基r2至r5形成,则这些更优选由r3和r4和/或r4和r5形成。优选残基r2至r5不形成环,并且更优选残基r2至r5中至多两个为甲基,其余为h。最优选r2至r5全部为h。此外,两个含氧环都优选地为饱和的或部分不饱和的或不饱和的。每个部分不饱和的或不饱和的含氧环可以具有一个或两个双键。更优选两个含氧环都是饱和的。优选的内部有机化合物的实例是2,2-二(2-四氢呋喃基)丙烷、2,2-二-(2-呋喃)-丙烷及其异构体或混合物。在最优选的实施例中,使用2,2-二(2-四氢呋喃基)丙烷(dthfp)与其异构体。dthfp典型地是d,l-(外消旋)-dthfp和内消旋-dthfp的1:1mol/mol非对映体混合物。根据本发明,已经发现使用富含dthfp的异构体的内部有机化合物,催化剂形态特性不受影响。已经发现通过使用富集的外消旋-dthfp,其中d,l-(外消旋)-dthfp/内消旋-dthfp的比例为至少2/1mol/mol,可以产生与等摩尔(外消旋)和(内消旋)混合物一样好的催化剂形态。dthfp的1h-nmr:外消旋和外消旋异构体的1:1mol/mol混合物在图1中披露。dthfp的1h-nmr:外消旋-dthfp(d,l-(外消旋)-dthfp)/内消旋-dthfp的2:1mol/mol混合物在图2中披露。通过与mgcl2络合,富集出人意料地成功。该富集步骤形成本发明的另外的发明步骤。根据本发明的方法,一个必要特征是,在用第13族金属化合物预处理mgcl2-mroh之前、期间或之后,但在用第4至6族过渡金属化合物处理它之前,将如上定义的内部有机化合物加入到催化剂混合物中。式(i)的内部有机化合物/加入到催化剂混合物中的加合物mgcl2*mroh的摩尔比为0,02-2,0mol/mol,优选为0,05至0,15mol/mol。因此,根据本发明的第一实施例,该固体催化剂组分是通过如下制备i)提供固体mgcl2*mroh载体,其中m为1至4,并且r为含有1至8个c原子的直链或支链烷基ii)用al化合物预处理步骤i)的固体载体颗粒iii)将式(i)的内部有机化合物加入到步骤ii)的经预处理的固体载体中,或iii’)与步骤ii)同时将式(i)的内部有机化合物加入到该固体载体中iv)用ticl4处理步骤iii)或iii’)的经预处理的固体载体颗粒,并且v)回收固体催化剂组分因此,根据本发明的第二实施例,该固体催化剂组分是通过如下制备i)提供固体mgcl2*mroh载体,其中m为1至4,并且r为含有1至8个c原子的直链或支链烷基ii-1)将式(i)的内部有机化合物加入到步骤i)的固体载体中iii-1)用al化合物预处理步骤ii-1)的固体载体颗粒iv-1)用ticl4处理步骤iii-1)的经预处理的固体载体颗粒,并且v-1)回收固体催化剂组分。根据上述实施例,可以在向载体中加入内部有机化合物之前或之后或者与加入内部有机化合物同时,将al化合物加入到固体载体中。最优选地,在第一和第二实施例中,m为2,7至3,3,roh为乙醇,铝化合物为三烷基铝化合物,例如三乙基铝,并且作为内供体使用2,2-二(2-四氢呋喃基)丙烷或2,2-二-(2-呋喃)-丙烷,尤其是2,2-二(2-四氢呋喃基)丙烷或者其异构体或混合物。根据本发明的催化剂制备方法,用第13族金属化合物,优选铝化合物进行预处理可以通过加入所述铝化合物在惰性有机溶剂中,优选在惰性脂族烃溶剂中,例如在庚烷中的溶液来进行。本发明的方法允许使用浓缩的铝化合物溶液。在使用三乙基铝(tea)的情况下,可以使用在惰性烃中的15wt-%至100wt-%的tea,优选在惰性脂族烃溶剂中(如在庚烷中)的25wt-%-100wt-%的tea溶液,或未搀水的tea。发现通过使用这些更浓缩的溶液,形态仍然是有利的,并且实现了废物的减少。最终固体催化剂组分的mg/timol/mol比为1至10,优选2至8,尤其是3至7,al/timol/mol比为0,01至1,优选为0,1至0,5,并且cl/timol/mol比为5至20,优选10至17。最终催化剂的mg组分优选仅来自固体mgcl2*mroh载体,即在催化剂制备中不使用另外的mg化合物。本发明的固体催化剂组分的颗粒在粒度上是均匀的,没有细粉或附聚物。本发明的进一步的益处是通过使用本发明的催化剂或通过本发明方法制备的催化剂,聚合物的分子量分布(mwd)可以变窄。此外,分子量的增加不以牺牲催化剂的生产率为代价。与使用相似类型的催化剂组分但使用不同内部有机化合物,和/或通过在ticl4处理步骤期间或之后加入内部有机化合物制备的,或使用所述有机化合物作为外部添加剂,或在沉淀的mgcl2基催化剂中使用所述有机化合物相比,生产率保持在可接受的高水平或甚至增加。因此,通过本发明的方法制备的催化剂的性能使得可以扩大聚乙烯的制备窗口,使得具有较高和较低量氢的聚合可以进行,同时保持良好的生产率。尤其是摩尔质量变化性、mwd、共聚单体响应、共聚单体组成分布(ccd)和活性和生产率的最佳组合使得本催化剂对于生产聚乙烯非常有吸引力。除了如上所述的固体催化剂组分之外,本发明的催化剂还包含也被称为活化剂的助催化剂。助催化剂是第13族金属的有机金属化合物,典型地为铝化合物。这些化合物包括烷基卤化铝,优选烷基氯化铝,例如乙基二氯化铝、二乙基氯化铝、乙基倍半氯化铝、二甲基氯化铝等。它们还包括三烷基铝化合物,例如三甲基铝、三乙基铝、三异丁基铝、三己基铝和三正辛基铝。也可以使用其他的烷基铝化合物,例如异戊二烯基铝。尤其优选的助催化剂是三烷基铝,其中特别使用三乙基铝、三甲基铝和三异丁基铝。本发明的催化剂还可以包含外部添加剂,如外部供体。可以使用的外部添加剂包括醚化合物,典型地为四氢呋喃、硅氧烷或硅烷型外部供体和/或烷基卤,如从现有技术中已知的。本发明的催化剂可用于聚合乙烯,任选地与一种或多种共聚单体一起。通常使用的共聚单体是优选选自c3-c20-α-烯烃,更优选选自c4-c10-α-烯烃的α-烯烃共聚单体,如1-丁烯、异丁烯、1-戊烯、1-己烯、4-甲基-1-戊烯、1-庚烯、1-辛烯、1-壬烯和1-癸烯,以及二烯,如丁二烯、1,7-辛二烯和1,4-己二烯,或环烯烃,如降冰片烯,及其任何混合物。最优选地,共聚单体是1-丁烯和/或1-己烯。本发明的催化剂允许生产宽范围的聚乙烯聚合物。因此,可以生产高密度、中密度和低密度乙烯聚合物。本发明的催化剂可以在用于生产聚乙烯的任何常用的单峰和多峰工艺中使用。典型地,聚乙烯聚合物是以多峰工艺配置生产的。可以在包含至少两个聚合阶段的本领域已知的任何合适的聚合工艺中生产多峰乙烯共聚物。优选以级联模式操作聚合阶段。聚合可以在浆料、溶液或气相条件下或其组合下操作。在wo-a-92/12182和wo-a-96/18662中披露了包含级联浆料和气相聚合阶段的合适工艺。在多峰聚合配置中,聚合阶段包含选自浆料和气相反应器的聚合反应器。在一个优选实施例中,多峰聚合配置包含至少一个浆料反应器,在一些实施例中为两个浆料反应器,随后为至少一个气相反应器,优选一个气相反应器。催化剂可以通过本领域已知的任何手段转移到聚合过程中。因此可以将催化剂悬浮在稀释剂中并将其保持为均匀的浆料。尤其优选的是使用粘度为20至1500mpa·s的油作为稀释剂,如wo-a-2006/063771中所披露的。还可以将催化剂与油脂和油的粘稠混合物混合并将所得糊料进料到聚合区。此外,可以以例如ep-a-428054中披露的方式使催化剂沉淀并将这样得到的催化剂泥浆的部分引入聚合区。浆料中的聚合通常在惰性稀释剂典型地为烃稀释剂如甲烷、乙烷、丙烷、正丁烷、异丁烷、戊烷、己烷、庚烷、辛烷等或其混合物中进行。优选地,稀释剂是具有1至4个碳原子的低沸点烃或这类烃的混合物。尤其优选的稀释剂是丙烷,可能含有少量的甲烷、乙烷和/或丁烷。浆料聚合中的温度典型地为从40℃至115℃,优选为从60℃至110℃,并且特别优选为从70℃至100℃。压力为从1至150巴,优选从10至100巴。浆料聚合可以在用于浆料聚合的任何已知的反应器中进行。这类反应器包括连续搅拌槽式反应器和环式反应器。尤其优选在环式反应器中进行聚合。任选地,如本领域已知的,将氢气进料到反应器中以控制聚合物的分子量。此外,可以将一种或多种α-烯烃共聚单体加入到反应器中以控制聚合物产物的密度和形态。这类氢和共聚单体进料的实际量取决于所得聚合物的所希望的熔体指数(或分子量)和密度(或共聚单体含量)。气相中的聚合可以在流化床反应器中,在快速流化床反应器中或沉降床反应器中,或在这些的任何组合中进行。典型地,流化床或沉降床聚合反应器在50℃-100℃,优选65℃-90℃的温度范围内操作。压力适宜为从10至40巴,优选从15至30巴。如果需要,也可以将一种或多种抗静电剂引入浆料和/或气相反应器中。该工艺还可以包含前反应器和后反应器。聚合步骤之前可以是预聚合步骤。预聚合步骤可以以浆料或气相进行。优选地,预聚合以浆料进行,并且尤其是在环式反应器中进行。预聚合步骤中的温度典型地为0℃-90℃,优选为20℃-80℃,并且更优选为30℃-70℃。压力不是关键的,并且典型地为从1至150巴,优选为从10至100巴。聚合可以连续进行或分批进行,优选地聚合连续进行。用于生产根据本发明的乙烯(共)聚合物的优选多阶段工艺包含浆料相聚合阶段和气相聚合阶段。每个阶段可以包含一个或多个聚合反应器。一种合适的反应器配置包含一至两个浆料反应器,优选环式反应器,以及一个气相反应器。这类聚合配置描述于例如专利文献中,例如北欧化工公司(borealis)的wo-a-92/12182和wo-a-96/18662中,并且被称为borstar技术。附图说明图1:dthfp的1h-nmr:d,l-(外消旋)-dthfp和内消旋-dthfp非对映体的1:1混合物。图2:dthfp的1h-nmr:d,l-(外消旋)-dthfp和内消旋-dthfp的2:1mol/mol混合物图3:对比实例1的催化剂的sem图图4:对比实例2的催化剂的sem图图5:对比实例4的催化剂的sem图图6:发明实例1的催化剂的sem图图7:发明实例2的催化剂的sem图图8:发明实例3的催化剂的sem图图9:发明实例4的催化剂的sem图实验部分方法熔体流动速率mfr2:190℃,2,16kg负荷mfr5:190℃,5kg负荷熔体流动速率根据iso1133测量,以g/10min表示。mfr指示聚合物的流动性,并因此指示聚合物的加工性。熔体流动速率越高,聚合物的粘度越低。分子量平均值,分子量分布(mn,mw,mz,mwd)根据iso16014-1:2003、iso16014-2:2003、iso16014-4:2003和astmd6474-12,通过凝胶渗透色谱法(gpc)测定分子量平均值(mz,mw和mn)、分子量分布(mwd)及由多分散指数pdi=mw/mn(其中mn是数均分子量,并且mw是重均分子量)描述的其宽度,使用了以下公式:对于恒定的洗脱体积间隔δvi,其中ai和mi是分别与洗脱体积vi相关的色谱峰片面积(chromatographicpeakslicearea)和聚烯烃分子量(mw),其中n等于从色谱图获得的积分限之间的数据点数。使用来自polymerchar公司(西班牙巴伦西亚)的配备红外(ir)检测器(ir4或ir5)的高温gpc仪器或来自安捷伦技术公司(agilenttechnologies)的配备有3xagilent-plgelolexis和1xagilent-plgelolexisguard柱的差示折射计(ri)。作为溶剂和流动相,使用用250mg/l2,6-二叔丁基-4-甲基-苯酚稳定的1,2,4-三氯苯(tcb)。色谱系统在160℃和1ml/min的恒定流速下操作。每次分析注入200μl样品溶液。使用安捷伦cirrus软件版本3.3或polymerchargpc-ir控制软件进行数据采集。使用通用校准(根据iso16014-2:2003),用19个窄mwd聚苯乙烯(ps)标准品在0,5kg/mol至11500kg/mol范围内校准柱设置。将ps标准品在室温下溶解若干小时。通过使用markhouwink方程和以下markhouwink常数来实现将聚苯乙烯峰分子量转换为聚烯烃分子量:kps=19×10-3ml/g,ηps=0.655kpe=39×10-3ml/g,ηpe=0.725kpp=19×10-3ml/g,ηpp=0.725使用三阶多项式拟合来拟合校准数据。所有样品制备浓度范围为0,5-1mg/ml,并且在连续轻轻摇动下,在160℃下,针对pp溶解2.5小时,或针对pe溶解3小时。熔体温度根据iso11357的差示扫描量热计(dsc),使用mettlerta820差示扫描量热计(dsc)在3±0,5mg样品上测量熔体温度。icp分析(al,mg,ti)催化剂的元素分析是通过取质量m的固体样品,在干冰上冷却来进行的。通过溶解在硝酸(hno3,65%,5%的v)和新鲜去离子(di)水(5%的v)中将样品稀释至已知体积v。将溶液进一步用di水稀释至最终体积v,并保持稳定2小时。使用空白(5%hno3溶液)和在5%hno3溶液中的0.5ppm、1ppm、10ppm、50ppm、100ppm和300ppm的al、mg和ti标准品校准的thermoelementalicap6300电感耦合等离子体-光学检测光谱仪(icp-oes),在室温下进行分析。在分析前一刻,使用空白和100ppm标准品“再取斜率(resloped)”,运行质量控制样品(在di水中的5%hno3溶液中的20ppmal、mg和ti)以确认再取的斜率。也在每5个样品后和计划的分析集结束时运行qc样品。使用285.213nm线监测mg的含量,并使用336.121nm线监测ti的含量。当icp样品中的al浓度在0-10ppm之间(仅校准为100ppm)通过167.079nm线监测铝的含量并且al浓度高于10ppm时,通过396.152nm线。报告的值是从相同样品获取的三个连续等分试样的平均值并通过将样品的原始质量和稀释体积输入到软件中得到的与原始催化剂相关。来自pe的共聚单体含量(ftir)使用nicoletmagna550ir光谱仪连同nicoletomnicftir软件基于傅里叶变换红外光谱(ftir)以已知的方式测定共聚单体含量。从样品压塑成型约220至250μm的薄膜。用具有已知含量的共聚单体的校准样品制备类似的薄膜。从薄膜的至少五个点测量厚度。然后用砂纸摩擦薄膜以消除反射。这些薄膜没有被未处理的(plain)手触摸以避免污染。对于每个样品和校准样品,制备至少两个薄膜。使用gracebyspecac薄膜压片机,在150℃下使用3+2分钟预热时间,1分钟压缩时间和4至5分钟冷却时间,从粒料压制薄膜。对于非常高分子量的样品,预热时间可能会延长或温度升高。共聚单体含量由波数约1378cm-1处的吸光度确定。校准样品中使用的共聚单体与样品中存在的共聚单体相同。通过使用2cm-1的分辨率,从4000至400cm-1的波数跨度和128的扫描次数进行分析。从每个薄膜中运行至少两个光谱。从1430至1100cm-1的波数范围的光谱确定共聚单体含量。通过选择所谓的短或长基线或两者,测量作为峰高的吸光度。短基线通过最小点绘制在约1410和1320cm-1之间,并且长基线绘制在约1410-1220cm-1之间。需要对每个基线类型进行特别校准。此外,未知样品的共聚单体含量需要在校准样品的共聚单体含量的范围内。从校准样品获得如下直线:其中ci是校准样品i的共聚单体含量a1378,i是样品i在约1378cm-1处的吸光度si是由校准样品i得到的薄膜的厚度k是校准线的斜率(通过回归分析得到),并且b是校准线的截距(通过回归分析得到)。通过使用如此获得的参数k和b,获得了样品的共聚单体含量其中cx是未知样品的共聚单体含量a1378,x是未知样品在约1378cm-1处的吸光度sx是由未知样品得到的薄膜的厚度k是从如上的校准样品获得的校准线的斜率b是从校准样品获得的校准线的截距。该方法给出以重量-%或摩尔%计的共聚单体含量,取决于校准中使用哪个。如果适当校准,也可以使用相同的方法来确定甲基数,即每1000个碳原子的ch3数。实例原料通过稀释来自科聚亚公司(chemtura)100%tea-s制备庚烷中的标准10wt%和25wt%tea(三乙基铝)溶液。从grace收到mgcl2*3etoh载体(表1)。由tcieuropen.v.提供2,2-二(2-四氢呋喃基)丙烷(dthfp),作为非对映异构体(d,l-(外消旋)-dthfp和内消旋-dthfp)的混合物(1:1)。由奥德里奇公司(aldrich)提供ticl4(金属杂质<1000ppm,金属分析>99.9%)。表1.mgcl2*3etoh载体。载体颗粒状20μm球形45μmmg(wt%)9,869,87乙醇(wt%)59,759,0etoh/mg(mol/mol)3,203,16d10(μm)12,132,1d50(μm)21,145,3d90(μm)31,863,8在下列实例中,披露了对比和发明催化剂的制备以及催化剂在聚合反应中的用途。表2中披露了催化剂和聚合物特性。图3-9中披露了催化剂的sem图对比实例1(ce1)按照ep0376936中描述的制备程序制备催化剂。a.经预处理的负载材料制备:在惰性气氛手套箱(<1ppmo2,h2o)中:将装备有两个橡胶隔片、温度计和机械搅拌器的干燥的250ml四颈圆底烧瓶用30ml庚烷和5g(20mmolmg)球形45μmmgcl2*3etoh载体填充。从手套箱中取出烧瓶,固定氮气入口和出口。将烧瓶置于冷却浴中,并以250rpm搅拌约10min。在1h时间内滴加预冷的在庚烷中的10wt%三乙基铝溶液(81.8g,72mmolal;al/etoh=1.1mol/mol),保持温度低于0℃(归因于c2h6的释放)。将所得悬浮液在20min内加热至80℃,并在该温度下以250rpm保持30min。允许该悬浮液在80℃下沉降5min,并通过插管除去上清液。将所得经预处理的负载材料伴随搅拌在80℃下用50ml甲苯洗涤一次,并且在50℃下用50ml甲苯洗涤两次(加入甲苯,以250rpm搅拌15min,沉降5min,通过插管除去液体)。b.催化剂制备:在室温下,将100ml甲苯加入到步骤a的经预处理的负载材料中。将混合物以250rpm搅拌约30min。逐滴添加未搀水的ticl4(3.984g,21.0mmol;ti/mg=1.0mol/mol),并且温度保持在25℃-35℃之间。将所得悬浮液在20min内加热至90℃,并在该温度下以250rpm保持60min。将该悬浮液在90℃下沉降5min,并通过插管除去液体。将所得催化剂在90℃下用50ml甲苯洗涤,在50℃下用50ml甲苯洗涤并在室温下用50ml戊烷洗涤(加入预热的甲苯或戊烷,以250rpm搅拌15min,沉降5min,通过插管除去上清液)。将催化剂在60℃下用氮气流干燥1.5h。产量为2g(70%,基于mg)。c.与1-丁烯的小型共聚在与1-丁烯共聚中测试催化剂(8.5mg)。三乙基铝(tea)用作助催化剂,al/ti摩尔比为15。聚合反应根据以下程序在3l小型反应器中进行:在20℃下将空的3l小型反应器用55ml的1-丁烯填充并在200rpm下搅拌。然后将1250ml丙烷作为聚合介质加入到反应器中,随后加入氢气(0.75巴)。将反应器加热至85℃,并且分批加入乙烯(3.7巴)。反应器压力保持在0.2巴的过压,并且搅拌速度增加到550rpm。将催化剂和助催化剂一起加入(催化剂和tea之间有几秒的预接触)具有另外100ml丙烷的反应器中。通过连续乙烯进料将总反应器压力保持在38.3巴。60min后,通过排出单体和h2来停止聚合。在称重前使获得的聚合物在通风橱中干燥过夜。d.聚合结果聚合反应的结果示于表2中。基于所生产的聚合物的量计算催化剂的活性。通过凝胶渗透色谱(gpc)测量分子量和分子量分布。通过ir测量丁烯-共聚单体含量。通过dsc测量共聚物的熔融温度。对比实例2(ce2)使用25wt%的tea和较低体积的溶剂制备催化剂。a.经预处理的负载材料制备:在惰性气氛手套箱(<1ppmo2,h2o)中:将装备有两个橡胶隔片、温度计和机械搅拌器的干燥的100ml四颈圆底烧瓶用30ml庚烷和5.1g(21mmolmg)球形45μmmgcl2*3etoh载体填充。从手套箱中取出烧瓶,固定氮气入口和出口。将烧瓶置于冷却浴中,并以250rpm搅拌约10min。在1h时间内滴加预冷的在庚烷中的25wt%三乙基铝溶液(30.4g,67mmolal;al/etoh=1.0mol/mol),保持温度低于0℃,归因于c2h6的释放。将所得悬浮液在20min内加热至80℃,并在该温度下以250rpm保持30min。将该悬浮液在80℃下沉降5min,并通过插管除去上清液。将所得经预处理的负载材料在室温下用50ml甲苯洗涤两次(加入甲苯,以250rpm搅拌15min,沉降5min,通过插管除去溶剂)。b.催化剂制备:在室温下,将50ml甲苯加入到步骤a的经预处理的负载材料中。将混合物以250rpm搅拌约30min。逐滴添加未搀水的ticl4(3.8g,20mmol;ti/mg=1.0mol/mol),并且温度保持在25℃-35℃之间。将所得悬浮液在20min内加热至90℃,并在该温度下以250rpm保持60min。将该悬浮液在90℃下沉降5min,并通过插管除去液体。将所得催化剂在90℃下用50ml甲苯洗涤两次,并且在室温下用50ml戊烷洗涤一次(加入预热的甲苯或戊烷,以250rpm搅拌15min,沉降5min,通过插管除去液体)。将催化剂在50℃下用氮气流干燥1.5h。产量为2.7g或基于mg的76%。c.与1-丁烯的小型共聚在与1-丁烯共聚中测试催化剂(6.2mg)。三乙基铝(tea)用作助催化剂,al/ti摩尔比为15。聚合反应根据对比实例1c中所述的程序在3l小型反应器中进行。d.聚合结果聚合反应的结果示于表1中。基于所生产的聚合物的量计算催化剂的活性。通过凝胶渗透色谱(gpc)测量分子量和分子量分布。通过ir测量丁烯-共聚单体含量。通过dsc测量共聚物的熔融温度。对比实例3(ce3)c.在用dthfp作为外部添加剂(dthfp/mg=0,14mol/mol,dthfp/ti=0,47mol/mol)的与1-丁烯的共聚中,测试了来自对比实例2的催化剂(6.2mg)。三乙基铝(tea)用作助催化剂,al/ti摩尔比为15。在聚合前1h将dthfp与手套箱中的tea溶液预混合。聚合反应根据对比实例1c中所述的程序在3l小型反应器中进行。d.聚合结果聚合反应的结果示于表2中。基于所生产的聚合物的量计算催化剂的活性。通过凝胶渗透色谱(gpc)测量分子量和分子量分布。通过ir测量丁烯-共聚单体含量。通过dsc测量共聚物的熔融温度。对比实例4(ce4)c.在与1-丁烯共聚中测试来自对比实例2的催化剂(7.2mg)。三乙基铝(tea)用作助催化剂,al/ti摩尔比为15。聚合反应根据以下程序在3l小型反应器中进行:在20℃下将空的3l小型反应器用70ml的1-丁烯填充并在200rpm下搅拌。然后将丙烷(1250ml)作为聚合介质加入到反应器中,随后加入氢气(0.40巴)。将反应器加热至85℃,并且加入一批乙烯(3.7巴)。反应器压力保持在0.2巴的过压,并且搅拌速度增加到550rpm。将催化剂和助催化剂一起加入(催化剂和tea之间有几秒的预接触)具有另外100ml丙烷的反应器中。通过连续乙烯进料将总反应器压力保持在37.5巴。60min后,通过排出单体和h2来停止聚合。在称重前使获得的聚合物在通风橱中干燥过夜。d.聚合结果聚合反应的结果示于表2中。基于所生产的聚合物的量计算催化剂的活性。通过凝胶渗透色谱(gpc)测量分子量和分子量分布。通过ir测量丁烯-共聚单体含量。通过dsc测量共聚物的熔融温度。发明实例1(ie1)在加入ticl4之前加入作为内部有机化合物的dthfp。a.经预处理的负载材料制备:在惰性气氛手套箱(<1ppmo2,h2o)中:将装备有两个橡胶隔片、温度计和机械搅拌器的干燥的100ml四颈圆底烧瓶用30ml庚烷和5g(20mmolmg)球形45μmmgcl2*3etoh载体填充。从手套箱中取出烧瓶,固定氮气入口和出口。将烧瓶置于冷却浴中,并以250rpm搅拌约10min。在1h时间内滴加预冷的在庚烷中的25wt%三乙基铝溶液(30.4g,67mmolal;al/etoh=1.1mol/mol),保持温度低于0℃。将所得悬浮液在20min内加热至80℃,并在该温度下以250rpm保持30min。将该悬浮液在80℃下沉降5min,并通过插管除去液体。将所得经预处理的负载材料在室温下用50ml甲苯洗涤两次(加入甲苯,以250rpm搅拌15min,沉降5min,通过插管除去液体)。b.催化剂制备:在室温下,向经预处理的负载材料中加入溶解在50ml甲苯中的dthfp(0.38g,dthfp/mg=0.1mol/mol)。将混合物在250rpm下搅拌约30min。逐滴添加未搀水的ticl4(3.8g,20mmol;ti/mg=1.0mol/mol),并且温度保持在25℃-35℃之间。将所得悬浮液在20min内加热至90℃,并在该温度下以250rpm保持60min。将该悬浮液在90℃下沉降5min,并通过插管除去液体。将所得催化剂在90℃下用50ml甲苯洗涤两次,并且在室温下用50ml戊烷洗涤一次(加入预热的甲苯或戊烷,以250rpm搅拌15min,沉降5min,通过插管除去液体)。将催化剂在50℃下用氮气流干燥1.5h。产量为3.3g或基于mg的89%。c.与1-丁烯的小型共聚在与1-丁烯共聚中测试催化剂(6.0mg)。三乙基铝(tea)用作助催化剂,al/ti摩尔比为15。聚合反应根据对比实例4c中所述的程序在3l小型反应器中进行。d.聚合结果聚合反应的结果示于表2中。基于所生产的聚合物的量计算催化剂的活性。通过凝胶渗透色谱(gpc)测量分子量和分子量分布。通过ir测量丁烯-共聚单体含量。通过dsc测量共聚物的熔融温度。发明实例2(ie2)在加入tea之前加入作为内部有机化合物的dthfp。该催化剂是基于45μmmgcl2*3etoh载体。a.经预处理的负载材料制备:在惰性气氛手套箱(<1ppmo2,h2o)中:将装备有两个橡胶隔片、温度计和机械搅拌器的干燥的100ml四颈圆底烧瓶用溶于30ml庚烷中的0.38gdthfp(dthfp/mg=0.1mol/mol)和5.1g(21mmolmg)球形45μmmgcl2*3etoh载体填充。从手套箱中取出烧瓶,固定氮气入口和出口。将烧瓶置于冷却浴中,并以250rpm搅拌约10min。在1h时间内滴加预冷的在庚烷中的25wt%三乙基铝溶液(30.4g,67mmolal;al/etoh=1.0mol/mol),保持温度低于0℃。将所得悬浮液在20min内加热至80℃,并在该温度下以250rpm保持30min。将该悬浮液在80℃下沉降5min,并通过插管除去液体。将所得经预处理的负载材料在室温下用50ml甲苯洗涤两次(加入甲苯,以250rpm搅拌15-120min,沉降5min,通过插管除去液体)。b.催化剂制备:在室温下,向经预处理的负载材料中加入50ml甲苯。将混合物在250rpm下搅拌约30min。逐滴添加未搀水的ticl4(3.8g,20mmol;ti/mg=1.0mol/mol),并且温度保持在25℃-35℃之间。将所得悬浮液在20min内加热至90℃,并在该温度下以250rpm保持60min。将该悬浮液在90℃下沉降5min,并通过插管除去液体。将所得催化剂在90℃下用50ml甲苯洗涤两次,并且在室温下用50ml戊烷洗涤一次(加入预热的甲苯或戊烷,以250rpm搅拌15min,沉降5min,通过插管除去液体)。将催化剂在50℃下用氮气流干燥1.5h。产量为3.6g(100%,基于mg)。c.与1-丁烯的小型共聚在与1-丁烯共聚中测试催化剂(5.7mg)。三乙基铝(tea)用作助催化剂,al/ti摩尔比为15。聚合反应根据对比实例4c中所述的程序在3l小型反应器中进行。d.聚合结果聚合反应的结果示于表2中。基于所生产的聚合物的量计算催化剂的活性。通过凝胶渗透色谱(gpc)测量分子量和分子量分布。通过ir测量丁烯-共聚单体含量。通过dsc测量共聚物的熔融温度。发明实例3(ie3)在加入tea之前加入作为内部有机化合物的dthfp。该催化剂是基于20μmmgcl2*3etoh载体。a.经预处理的负载材料制备:在惰性气氛手套箱(<1ppmo2,h2o)中:将装备有两个橡胶隔片、温度计和机械搅拌器的干燥的100ml四颈圆底烧瓶用溶于30ml庚烷中的0.38gdthfp(dthfp/mg=0.1mol/mol)和5g(20mmolmg)颗粒状20μmmgcl2*3etoh载体填充。从手套箱中取出烧瓶,固定氮气入口和出口。将烧瓶置于冷却浴中,并以250rpm搅拌约10min。在1h时间内滴加预冷的在庚烷中的25wt%三乙基铝溶液(30.4g,67mmolal;al/etoh=1.0mol/mol),保持温度低于0℃。将所得悬浮液在20min内加热至80℃,并在该温度下以250rpm保持30min。将该悬浮液在80℃下沉降5min,并通过插管除去液体。将所得经预处理的负载材料在室温下用50ml甲苯洗涤两次(加入甲苯,以250rpm搅拌15-120min,沉降5min,通过插管除去液体)。b.催化剂制备:在室温下,向经预处理的负载材料中加入50ml甲苯。将混合物在250rpm下搅拌约30min。逐滴添加未搀水的ticl4(3.8g,20mmol;ti/mg=1.0mol/mol),并且温度保持在25℃-35℃之间。将所得悬浮液在20min内加热至90℃,并在该温度下以250rpm保持60min。将该悬浮液在90℃下沉降5min,并通过插管除去液体。将所得催化剂在90℃下用50ml甲苯洗涤两次,并且在室温下用50ml戊烷洗涤一次(加入预热的甲苯或戊烷,以250rpm搅拌15min,沉降5min,通过插管除去液体)。将催化剂在50℃下用氮气流干燥1.5h。产量为3.4g(94%,基于mg)。c.与1-丁烯的小型共聚在与1-丁烯共聚中测试催化剂(7.0mg)。三乙基铝(tea)用作助催化剂,al/ti摩尔比为15。聚合反应根据对比实例4c中所述的程序在3l小型反应器中进行。d.聚合结果聚合反应的结果示于表2中。基于所生产的聚合物的量计算催化剂的活性。通过凝胶渗透色谱(gpc)测量分子量和分子量分布。通过ir测量丁烯-共聚单体含量。通过dsc测量共聚物的熔融温度。发明实例4(ie4)在加入tea之前加入作为内部有机化合物的富集的外消旋-dthfp(d,l-(外消旋)-dthfp/内消旋-dthfp=2/1mol/mol)。该催化剂是基于45μmmgcl2*3etoh载体。在室温下,将无水mgcl2(0.95g,10mmol)与3.68gdthfp(20mmol;d,l-(外消旋)-dthfp/内消旋-dthfp=1/1mol/mol)混合。将所得悬浮液加热至130℃并搅拌3h。将混合物冷却至室温并过滤。所得滤液含有d,l-(外消旋)-dthfp和内消旋-dthfp(2/1mol/mol;产率93%)的混合物,并且照原样用于催化剂合成。a.经预处理的负载材料制备:在惰性气氛手套箱(<1ppmo2,h2o)中:将装备有两个橡胶隔片、温度计和机械搅拌器的干燥的100ml四颈圆底烧瓶用溶于30ml庚烷中的0.38g富集的外消旋-dthfp(dthfp/mg=0.1mol/mol)和5.1g(21mmolmg)球形45μmmgcl2*3etoh载体填充。从手套箱中取出烧瓶,固定氮气入口和出口。将烧瓶置于冷却浴中,并以250rpm搅拌约10min。在1h时间内滴加预冷的在庚烷中的25wt%三乙基铝溶液(30.4g,67mmolal;al/etoh=1.0mol/mol),保持温度低于0℃。将所得悬浮液在20min内加热至80℃,并在该温度下以250rpm保持30min。将该悬浮液在80℃下沉降5min,并通过插管除去液体。将所得经预处理的负载材料在室温下用50ml甲苯洗涤两次(加入甲苯,以250rpm搅拌15-120min,沉降5min,通过插管除去液体)。b.催化剂制备:在室温下,向经预处理的负载材料中加入50ml甲苯。将混合物在250rpm下搅拌约30min。逐滴添加未搀水的ticl4(3.8g,20mmol;ti/mg=1.0mol/mol),并且温度保持在25℃-35℃之间。将所得悬浮液在20min内加热至90℃,并在该温度下以250rpm保持60min。将该悬浮液在90℃下沉降5min,并通过插管除去液体。将所得催化剂在90℃下用50ml甲苯洗涤两次,并且在室温下用50ml戊烷洗涤一次(加入预热的甲苯或戊烷,以250rpm搅拌15min,沉降5min,通过插管除去液体)。将催化剂在50℃下用氮气流干燥1.5h。产量为3.8g(100%,基于mg)。c.与1-丁烯的小型共聚在与1-丁烯共聚中测试催化剂(7.2mg)。三乙基铝(tea)用作助催化剂,al/ti摩尔比为15。聚合反应根据对比实例4c中所述的程序在3l小型反应器中进行。d.聚合结果聚合反应的结果示于表2中。基于所生产的聚合物的量计算催化剂的活性。通过凝胶渗透色谱(gpc)测量分子量和分子量分布。通过ir测量丁烯-共聚单体含量。通过dsc测量共聚物的熔融温度。表2.催化剂和聚合结果*测试共聚条件:t=85℃,c2=5mol%,h2/c2=40mol/kmol,c4/c2=770mol/kmol,t=1h,al/ti=15mol/mol。**测试共聚条件:t=85℃,c2=5mol%,h2/c2=20mol/kmol,c4/c2=970mol/kmol,t=1h,al/ti=15mol/molnd-未定义从结果可以看出,通过本发明方法生产的聚合物的分子量与对比实例中的相比更高或至少在相同的水平,而且活性仍保持在良好的水平。同时,mwd较窄,共聚单体含量较低,并且熔融温度低于对比实例。本发明实例的催化剂的形态也是均匀的。在实例ce4中,活性和mw处于良好的水平,然而,其他特性(mwd,c4含量和mp)不利地比本发明实例中的更高。此外,催化剂的形态一点也不令人满意,表明在大规模生产中可能存在的困难。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1