由右旋糖制备丙烯酸的方法与流程
本发明总体上涉及制备生物基丙烯酸的方法,并且更具体地,涉及从糖制备生物基丙烯酸的方法。
背景技术:
丙烯酸是一种有价值的工业商品并且具有多种用途。由丙烯酸制成的聚合物用于制造胶粘剂、粘合剂、涂料、油漆、抛光剂和超吸收性聚合物,后者进而用于例如包括尿布和卫生产品的一次性吸收制品中。
目前丙烯酸是由石油源材料制成。例如,长期以来丙烯酸通过丙烯的催化氧化制备。然而,近年来,随着对开发用于制造丙烯酸和其他常规石油化学品的基于可再生源的工艺的需求日益增长的意识,大量的研究已经致力于鉴定和开发用于从可再生资源制备丙烯酸的方法。
因此,大量的参考文件描述了用于将甘油转化成丙烯酸和/或丙烯酸酯的方法,这些方法通常利用甘油例如在从植物油制造生物柴油(脂肪酸甲酯)中生产的甘油,参见,例如,授予dubois等人的us7,396,962以及其中引用的参考文献。
与本发明的方法更直接相关,同样已经做了许多努力来开发用于从碳水化合物和/或碳水化合物衍生的原料制造丙烯酸的方法。可以衍生自碳水化合物并且已经严谨地评估过的一种原料是3-羟基丙酸、或3-hpa。授予holmen的us2,859,240(1958)指出3-hpa的脱水是一种“比较简单并且经济的方法”,但是总结出“起始材料既不是成本上低的也不是数量上易于获得的”(第1栏,第55-58行)45年后提供了基本上相同的评估,其中在kumar等人的“3-羟基丙酸的生物生产的研究进展(recentadvancesinbiologicalproductionof3-hydroxypropionicacid)”生物技术进展(biotechnologyadvances),第31卷,第945-961页(2013)中,作者总结出尽管在过去的十年朝向“商业生产...在不久的将来”的“重大进展”,“很多重要的问题仍然存在并且要求更加广泛的研究”。
可以衍生自碳水化合物并且也已经是大量研究的主题的另一种原料为乳酸。在同一1958holmen专利中,例如,指出乳酸由于其随时可用性有一段时间被认为比3-hpa优选作为一种预期原料,(参考1950的到那时对于开发用于将乳酸和乳酸的低级烷基酯转化为丙烯酸和相应的丙烯酸的低级烷基酯的方法的努力的综述)。一种商业上可行的方法对于将乳酸转化为丙烯酸的仍然还保持是难以实现的,如通过最近提交的许多正在进行的专利申请所证明的。
授予ozmeral等人的wo2012/033845、授予dongare等人的wo2012/156921以及授予lingoes等人的wo2013/155245是开发用于将乳酸(和/或乳酸酯的酯)转化为丙烯酸(和/或相应的丙烯酸酯的酯)的商业上可行的方法的这些正在进行的努力的代表,并且各自进而综述了相当大量的另外公开技术的主要部分(详述针对同一目的的先前的工作)。
在wo2012/033845中,一种含有乳酸铵的发酵培养液被描述为根据生产丙烯酸酯的四种途径之一进行加工。在第一途径中,首先从该发酵培养液中纯化乳酸。然后使高度纯化的乳酸在高温下并且在一种适当的催化剂存在下经受气相脱水反应以便生产丙烯酸,将该丙烯酸进而在一种酯化催化剂存在下酯化以便提供丙烯酸酯。在第二途径中,使该发酵培养液中的乳酸“无需大量纯化下”脱水,接着进行酯化反应以便生产丙烯酸酯。在第三途径中,使在该发酵培养液中的乳酸铵经受同时的脱水和酯化反应以便产生一种丙烯酸酯产物,而在第四途径中,使在该发酵培养液中的乳酸铵无需大量纯化下首先经受一种酯化反应以便产生乳酸酯,并且然后使这种乳酸酯脱水以便提供一种丙烯酸酯产物。在根据这个第四途径一个“最优先的”实施例中,使一种含有乳酸铵的发酵培养液通过蒸发水来浓缩并且经受与c1-c10烷基醇的酯化,优选在不存在任何外来的酯化催化剂下。在浓缩过程期间释放的氨被捕获用于再循环至该乳酸发酵中,连同在酯化反应的过程中释放的另外的氨。然后使在该第一阶段中获得的乳酸酯脱水以便产生一种相应的丙烯酸酯。
在授予dongare等人的wo2012/156921中,提供了一种具有从乳酸到丙烯酸的改进的选择性以及降低的乙醛和其他产物生产的催化剂用于乳酸到丙烯酸的脱水,该催化剂包括一种如任选地用5重量百分比的钠改性的磷酸钙(以从1.5至1.9的钙与磷酸根的比)。该方法被描述为涉及在一个固定床反应器中在高纯的氮气下在370至380摄氏度的温度下预热该催化剂从20至40分钟,然后使50-80wt百分比预热的乳酸溶液的蒸汽借助于一种氮气载气穿过石英固定催化剂床反应器。所报告的在这些条件下的乳酸转化率是100百分比,其中60至80百分比的丙烯酸选择性和15-35百分比的乙醛选择性。
在授予lingoes等人的wo2013/155245中,首先参考与dongare等人中报道的特性类似的特性的多个团体的研究,该研究证实磷酸盐和硝酸盐盐可以令人希望地改变酸性催化剂的表面酸度以便抑制特别是乳酸至乙醛的脱羰/脱羧。
lingoes等人主张即使具有对乙醛的降低的选择性,然而甚至减少的量是有问题的,因为副产物可能沉积在该催化剂上并且导致结垢和过早且迅速的催化剂失活。进一步,一旦沉积,这些副产物可以催化其他不希望的反应,例如聚合反应(第0005段)。
同样,除了所讨论的沉积在催化剂上造成的困难之外,lingoes等人指出以下困难:甚至非常少量的副产物如乙醛、丙酸、一氧化碳、二氧化碳、2-3-戊二酮和乳酸低聚物可以在根据那时已知的乳酸到丙烯酸方法加工丙烯酸以制造超吸收性聚合物中产生,使得现有文献的显著主体围绕从该丙烯酸去除这些杂质。
lingoes等人,引用文献us6,541,665和美国公开专利申请2011/0257355作为此文献主体的范例。在us6,541,665中,一种5-阶段结晶(含有两个纯化阶段和三个汽提阶段)对于获得99.94%的丙烯酸是有效的,该丙烯酸含有除了其他种类按重量计百万分之2600的乙酸和358ppm的丙酸。在us2011/0257355中,描述了一种以单次结晶从衍生自甘油脱水/氧化的粗反应混合物中去除丙酸以获得99%丙烯酸的方法。根据lingoes等人,在他们的改进的催化剂和方法之前,用于将乳酸转化成丙烯酸的先前技术方法产生的副产物的量值太高(“极其高”)以致于甚至不能利用此类纯化方法。
技术实现要素:
一方面,本发明涉及一种从右旋糖制备丙烯酸的方法,该方法包括:
a)在生物催化剂存在下发酵右旋糖以产生含有乳酸的发酵培养液;
b)从该发酵培养液中去除固体以产生澄清的发酵培养液;
c)通过萃取到有机溶剂中从该澄清的发酵培养液中去除乳酸;
d)在从其中去除乳酸后,从该发酵培养液剩余部分中分离该乳酸负载的有机溶剂;
e)将该发酵培养液剩余部分的至少一部分再循环至该发酵步骤中;
f)使该乳酸负载的溶剂中的乳酸与氨反应以提供包含乳酸铵的粗脱水进料;
g)将该粗脱水进料中的乳酸铵与有机溶剂进行分离以提供脱水进料;
h)进行该脱水进料中的乳酸铵的气相脱水以产生粗丙烯酸产物;
i)通过以下方法纯化该粗丙烯酸产物以提供纯化的丙烯酸产物,该方法包括:
第一蒸馏以去除乙醛和氨塔顶馏出物,并提供主要由丙烯酸和丙酸组成的塔底物流,并且
来自该第一蒸馏的塔底物流的第二蒸馏以提供富含丙烯酸的第二蒸馏塔顶馏出物流和富含丙酸的第二蒸馏塔底物流;
并且,
j)通过熔融结晶、色谱法或熔融结晶和色谱法两者进一步纯化该第二蒸馏塔顶馏出物流中的丙烯酸。
在一个实施例中,该纯化的丙烯酸产物至少具有可接受的纯度以作为冰丙烯酸被商业销售。
在另一实施例中,该纯化的丙烯酸产物含有按重量计小于3000ppm的丙酸。
在另一实施例中,该纯化的丙烯酸产物含有按重量计小于1000ppm的丙酸。
在根据本发明的方法的另一个实施例中,该方法进一步包括在合适的催化剂存在下进行第二蒸馏塔底物流中丙酸的氧化脱氢以提供附加的丙烯酸。然后这种附加的丙烯酸可以通过熔融结晶、色谱法或熔融结晶和色谱法两者进行纯化,视情况而定,考虑到任何未转化的剩余的丙酸和对于实现所希望的冰丙烯酸产物可用的丙酸极限(由于来自常规石油衍生原料的冰丙烯酸的制造商和购买者的纯度要求确实有所不同)。典型地,虽然不一定,但这将至少部分地通过以下进行:从第二蒸馏塔底流中的丙酸的氧化脱氢中再循环丙烯酸,以与第二蒸馏塔顶馏出物流中的丙烯酸组合,之后通过熔融结晶、色谱法或熔融结晶和色谱法两者进行其纯化。
在另一个实施例中,该方法进一步包括在合适的催化剂的存在下,用氢气源进行第二蒸馏塔底物流中的丙烯酸的氢化以从该第二蒸馏塔底物流产生商业质量的丙酸副产物。
附图说明
图1a是在一个实施例中根据本发明的方法的一部分的示意图。
图1b是在一个替代实施例中根据本发明的方法的一部分的示意图。
图2是在一个实施例中根据本发明的方法的第二下游部分的示意图。
图3描绘了脉冲测试的结果,该测试例如使用水作为洗脱液,在图1和图2中示意性描绘的方法中使用两性树脂用于进行从丙烯酸产物中的过量丙酸的色谱分离。
图4描绘了相同的树脂体系的脉冲测试的结果,但是使用5%丙酮在水中的混合洗脱液。
图5描绘了使用甲醇共溶剂而不是丙酮的脉冲测试的结果。
图6描绘了使用更高百分比的甲醇共溶剂的脉冲测试的结果。
图7示意性地描绘了以下基于初始脉冲测试的某些实例中所使用的12-柱模拟移动床色谱装置。
具体实施方式
现在转到图1a/1b和图2,根据本发明的方法的一个说明性实施例以两部分示意性地示出,在图1a和1b中示出了根据本发明的整体方法的第一部分的两种可能的配置。图1a和1b描绘了用于连续产生粗丙烯酸产物流的方法的第一上游部分的替代配置,而图2描绘了用于净化粗丙烯酸产物流的第二下游部分,由此可以连续生产商业上可接受的冰丙烯酸产物。
现在转到如图1a中的一个实施例(10)示出的方法的上游部分,将右旋糖12与微生物14和微生物14的营养物16供应到发酵罐18,其中右旋糖生物转化为呈含乳酸的发酵培养液20的形式的乳酸。
用于提供含乳酸的发酵培养液的右旋糖的发酵是商业上实践的,并且本领域技术人员将熟悉可用于在发酵罐中由右旋糖生产乳酸的许多微生物和相关方法。合适方法的实例包括在hara等人的us2012/0214214(使用粟酒裂殖酵母的抗酸转化体)、sineokij等人的ru2268304c1(使用粟酒裂殖酵母的重组菌株)和liu等人的us2005/0112737(使用包含包括外源性乳酸脱氢酶基因的基因组的耐酸酵母菌株)中描述的那些方法,所有这些都通过引用并入本文。
在说明性实施例中,将含乳酸的发酵培养液20收集在乳酸培养液罐22中。如通常在发酵培养液的加工领域中常规的,固体去除步骤23然后从含乳酸的发酵培养液20中去除固体,以提供例如细胞碎片已经被去除(如由参考号25示意性表示的)的澄清的含乳酸的发酵培养液24。各种手段在用于去除固体的发酵培养液的加工领域中是众所周知的,并且因此可用于固体去除步骤23中,包括但不限于各种形式的过滤、絮凝、沉降、离心及类似物,然而在优选的实施例中使用超滤。
将澄清的发酵培养液24在任何情况下都连续地供应到用于将乳酸从发酵培养液24中去除到合适的有机溶剂中的溶剂萃取步骤26,同时将25中回收的细胞体再循环到发酵罐18中。在一个实施例中,溶剂萃取步骤26涉及使用以壳管式构型安排的多个中空纤维膜,尽管已知并且可以选择使用许多不同的膜构型。例如,可以使用平面片材膜或平面片材膜堆叠、或以螺旋构型安排的多个同心管状膜(通常称为螺旋过滤器)。本领域的且熟悉基于膜的气体回收或分离系统的技术人员将能够很好地选择合适的膜系统和构型,但是目前优选的实施例将采用亲水性纳滤膜。如以下实施例所示,由德国伍珀塔尔(wuppertal,germany)的迈博锐公司(membranagmbh)出售的liqui-celtm膜接触器中使用的疏水膜的类型,也可以使用但现在不太优选。
在使用以壳管式构型安排的中空纤维膜的一个实施例中,将已经在溶剂罐30中加入氢氧化铵28的有机溶剂经由流32供应到用于步骤26的中空纤维膜的壳程。来自含乳酸的水性进料24的乳酸沿着并且径向穿过中空纤维膜移动,以在溶剂中在壳程上形成乳酸铵。然后进料24的乳酸耗尽的剩余部分可以优选地至少部分经由流34a再循环以利用其中所含的额外的营养物来支持发酵罐18中的发酵,任何不被如此使用的乳酸耗尽的剩余部分如所示经由流34b再循环到乳酸培养液罐22,如果没有为了在乳酸培养液罐22中和在含乳酸的水性进料24中维持所需的乳酸浓度所需的清洗部分36。
将乳酸铵同时经由流38在溶剂中供应到沉降槽40中,其中乳酸铵通过重力沉降浓缩并部分地从有机溶剂中分离出来。在任选的添加步骤中,在沉降槽40之前,残留的阴离子物质(例如磷、硫、铝和铁)和可能已经随着乳酸转移到有机溶剂中的颜色体可以通过用吸附介质的吸附和/或离子交换或排斥中的一种或多种,根据已知的方法和使用常规的技术来移除。然后将来自沉降槽40底部的乳酸铵溶液42与蒸发器44连通,用于将蒸气态的乳酸铵进料46供到脱水反应器48,同时回收的溶剂从沉降槽40的顶部在流50中再循环以再利用。如所指示的从蒸发器44中取出少量清洗流52,以维持在蒸气态的乳酸铵进料中的乳酸铵浓度在希望的范围内。
在反应器48中,将蒸气态的乳酸铵进料46中的乳酸铵脱水成包括丙烯酸和少量其它副产物(例如丙酸、乙醛、一氧化碳和二氧化碳)的产物。可以考虑将各种脱水催化剂和相关方法用于反应器48中,但是在一个实施例中,使用水性无机碱处理的磷酸铝催化剂(如paparizos等人的us4,786,756中所描述的),此专利现在通过引用并入本文。在paparizos等人中,乳酸和/或乳酸铵通过以下方式转化为气相中的丙烯酸:使水和乳酸和/或乳酸铵的混合物在每摩尔乳酸和/或乳酸铵从0.1至50、通常0.5至50摩尔的蒸汽下与磷酸铝接触,该磷酸铝已经用水性无机碱处理并在从300摄氏度至650摄氏度(通常从450至550摄氏度)的范围内的温度下煅烧从10分钟至20小时,通常从30分钟至10小时。该反应在从250至500摄氏度、通常从320至375摄氏度的温度下,以及0.1至15秒、通常2至4秒的接触时间下进行。当乳酸铵脱水时,产生乳酸和氨,如果需要,氨可以在右旋糖发酵成乳酸中被用作营养物。参考图1,流34a因此可以至少部分地再循环到发酵罐18中,未在萃取膜单元26中与乳酸反应并穿过该膜进入含乳酸的水性发酵培养液20中的任何氨为在发酵罐18中进行的发酵提供附加的营养物。
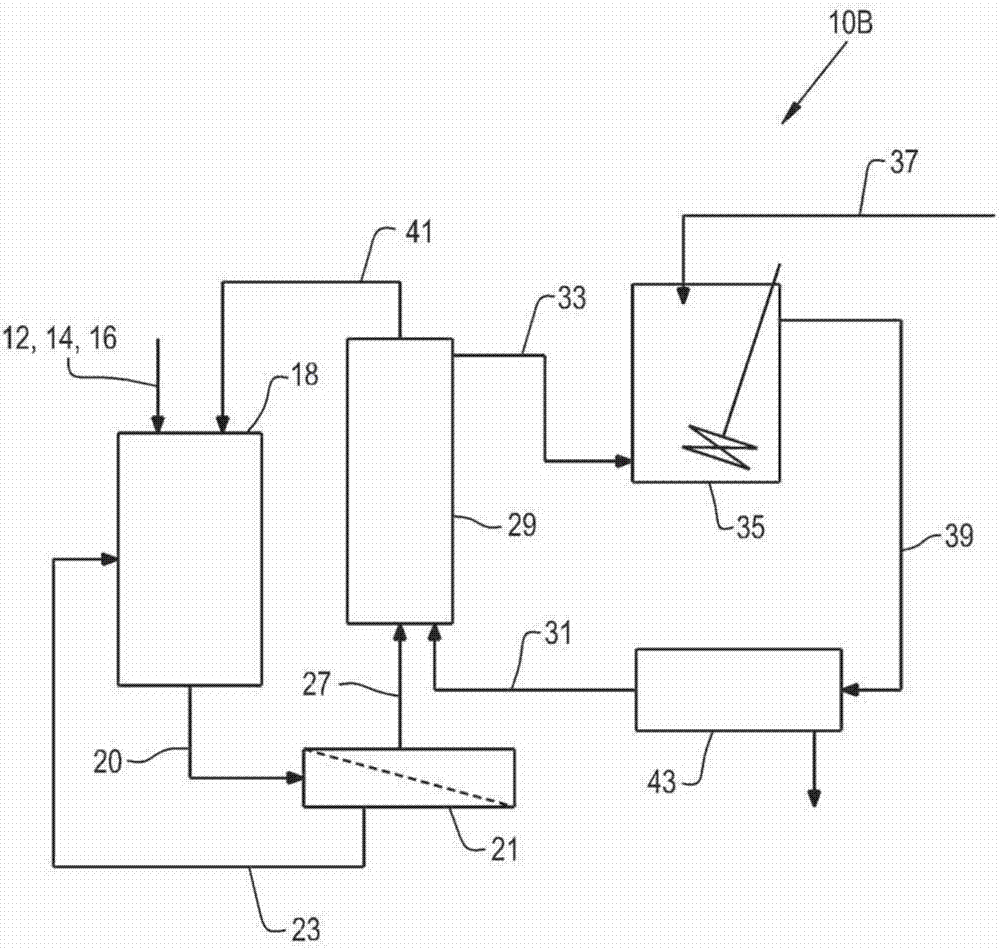
图1b描绘了用于连续产生粗丙烯酸产物流的方法的第一上游部分的替代配置。在一个实施例10b中,如图1a中将右旋糖、微生物和微生物的营养物12、14和16供应到发酵罐18,其中右旋糖生物地转化为呈含乳酸的发酵培养液20的形式的乳酸。
发酵培养液20在超滤步骤21中经历超滤,产生返回到发酵罐18的细胞体的再循环流23和供应给溶剂萃取步骤29的澄清的发酵培养液27。在溶剂萃取步骤29中,将有机溶剂31与澄清的发酵培养液27紧密混合以从其中萃取乳酸,或更优选地在其中使用亲水性纳滤膜材料允许乳酸从澄清的发酵培养液中去除进入该有机溶剂,同时基本上防止有机溶剂进入发酵培养液以及来自发酵培养液的较高分子量的颜色体与乳酸一起进入有机溶剂。如前所述,可以以在普通专业人员的技能范围内的各种已知的空间构型使用各种亲水性纳滤膜材料。
含有萃取的乳酸(33)的有机溶剂流然后行进到容器35,其中在水性氢氧化铵流37中供应的氨与萃取的乳酸反应形成乳酸铵产物39,而乳酸已经被去除的发酵培养液剩余部分41再循环回到发酵罐18中。
然后将乳酸铵产物39在容器43中相分离以提供乳酸铵水溶液45,然后将该乳酸铵水溶液以在图1a的实施例中流42的方式供给到蒸发器44中以在反应器48中经受气相脱水。含有再生的有机溶剂的有机相然后经由流31再循环(根据需要使用任何额外的补充溶剂),以进一步用于从另外的澄清发酵培养液27中回收乳酸。
现在参考如图2中示意性示出的本发明方法的说明性实施例的第二部分,在反应器48中完成的脱水产生粗丙烯酸产物50,该粗丙烯酸产物包含丙烯酸、丙酸、乙醛、氨、二氧化碳和一氧化碳以及相当数量的水。通过在萃取塔54中将有机物从粗丙烯酸产物50中萃取到合适的逆流流动的萃取剂52(例如丙烯酸乙酯)中,将大部分的该水从从粗丙烯酸产物50的剩余部分分离出来。如图2示出,在更轻的有机组分(氨、一氧化碳和二氧化碳)在流58中从后续的闪蒸容器60中闪蒸出来之前,过量的水经由流56被去除。将呈第一蒸馏塔进料62形式的剩余部分在第一蒸馏塔64中蒸馏,以优选去除流66中除了丙烯酸和丙酸之外的基本上所有残留的较轻组分(例如氨、乙酸、甲酸和乙醛)塔顶馏出物,而将主要且优选基本上完全由丙烯酸和丙酸组成的底部料流68进料到在非常低的压力(例如,大约10kpa(0.1巴))下操作的第二蒸馏塔70中,以通过单独的蒸馏优选实现尽可能实现的那样完全地分离在粗丙烯酸产物50中的丙烯酸和丙酸。
由于丙烯酸和丙酸的沸点彼此非常接近,所以在所示的实施例中,将含有来自粗丙烯酸产物50的大部分所需丙烯酸的第二蒸馏塔顶物流72仍然传递到进一步的纯化装置。在一个实施例中,该进一步的纯化方法将如共同转让给schultz等人的wo2015/031182中所述,其通过引用并入本文。因此,在一个实施例中(以下几个实施例涉及的),色谱法,特别是模拟移动床色谱法,用于从第二蒸馏塔顶馏出物流72中分离出过量的丙酸,优选达到由此产生冰丙烯酸品质产品的程度。在另一个实施例中,色谱法与结晶组合使用用于分离出过量的丙酸,并提供优选具有冰丙烯酸纯度的丙酸减少的、生物基丙烯酸产物。
连续工业规模的吸附方法由于它们的效率是熟知的。一种连续逆流移动床色谱装置的运行特别增强了传质驱动力,从而相比于传统的分批洗脱色谱法对于给定量的吸附剂允许更高的处理通过量和所希望的组分的更完全的分离。然而,在这种逆流运行模式中流体相和固相二者必须处于运动中。然而,固体的运动呈现出相当大的技术问题,包括侵蚀该吸附剂(产生导致高的压降的细粉)和设备磨损。因为这些困难,已经开发了模拟移动床色谱系统,其中该固体吸附剂是保持静态但是进行在流体流动方向上的所有入口流作为出口流的周期性单柱移动(one-columnshift)。以此方式,相对于该流体流动产生固体的表观或模拟逆流运动。此类模拟移动床色谱系统被广泛用于多种工业和各种各样的应用中,并且是优选的方法,其中色谱法用于从例如来自第二蒸馏塔70的塔顶馏出物流72中去除过量的丙酸,并提供丙酸含量减少的丙烯酸产物(含有优选按重量计小于3000ppm的丙酸,以及更优选按重量计小于1000ppm的丙酸)。
模拟移动床色谱系统的一种详细处理,这些系统的设计和运行不需要在本文中进行,因为这些系统在使用中并且是熟知的;然而,本领域的普通技术人员可以如所希望的在公开文献中找到额外的信息,例如,在gomes和rodrigues,“simulatedmovingbedchromatography:fromconcepttoproof-of-concept”,chemicalengineeringtechnology,vol.35,no.1,pp17-34(2011),[“模拟移动床色谱法:从原理到概念验证”,化学工程技术,第35卷,第1期,第17-34页(2011)],该文章特此通过引用结合在此,并且将由下面所描述的实例来指导。
刚刚引用的实例示出两性树脂–包括附接到聚苯乙烯基质上的阳离子和阴离子官能团二者–对于我们的应用是有效色谱树脂。这些树脂典型地用于分离一种电解和非电解质,或用于分离两种电解质。除了由三菱化学公司(mitsubishichemical)出售并且用于以下的若干实例中的diaionamp-03两性离子交换树脂之外,多种两性色谱树脂是可商购的并且可以使用的。例如,同一树脂的一种较早的形式以diaionamp-01商品名出售并且在一定程度上仍然可能是可商购的;尽管据报道具有不同和可能较不均匀的珠粒大小,该树脂的这个较早形式也应该适用于在方法步骤16中使用。
该diaionamp-03两性离子交换树脂本身由其供应商描述为一种两性离子交换树脂(其中一个季铵基团和一个羧基被结合在交联的聚苯乙烯框架上)、具有260μm的均匀的珠粒尺寸和出色的降解和浸出耐受性。建议的应用使用水作为洗脱液(流动相)以分离在水溶液中的不同的盐;因此预期的是在一个替代的实施例中,在塔顶馏出物流72中的丙酸和丙烯酸可以使用该diaionamp-03两性离子交换树脂或一种类似的两性树脂通过以下方式进行分离:从该丙酸和丙烯酸形成丙酸酯和丙烯酸酯并且然后分离这些酯。
使用水作为洗脱液(如由三菱公司建议用于分离盐)可能会需要大量的水,如通过脉冲测试所示,该脉冲测试的结果示于图3中,因为在图3中明显的丙烯酸的保留时间并且丙烯酸峰的轻微拖尾。优选地,于是该洗脱液是水与一种或多种有机溶剂的组合。甲醇和丙酮二者被证明在减少丙烯酸峰的保留时间和在降低总的洗脱要求上是有效的(如通过图4-6所示),虽然本领域技术人员将能够很好地识别实现这些目的的其他有机溶剂,并且在以下实例的方式之后优化它们与水的使用。
在其他实施例中,也可以通过色谱法和结晶的组合去除过量的丙酸。使用熔融和分步结晶二者用于纯化丙烯酸是非常熟知并且已经建立的,并且多种动态、悬浮和静态结晶方法和装置是已知的。熔融结晶基本上根据初始体系的热力学平衡通过冷却和结晶所希望的产物通过从熔体分离化合物而操作,并且在本发明的上下文中,用于产生相比于进料到结晶器的含有丙酸的丙烯酸的溶液以及保留丙酸在溶液中的母液具有丙酸含量减少的丙烯酸。
认为的是可以使用任何已知的结晶器,并且其类型或尺寸不受特别限制。例如,由瑞士温特图尔苏尔寿有限公司(sulzerltd.,winterthur,switzerland)出售的类型的降膜结晶器是目前用于纯化丙烯酸的一种类型的动态层结晶装置并且在一个实施例中可以用于图2的具体实施例中描绘的数个熔融结晶阶段,尽管授予dubois的us8,440,859表达了对于一系列的降膜结晶器接着是一个最终静态结晶器的优选。在大多数降膜结晶器中,纯化的丙烯酸在管的内表面上结晶,但一种降膜结晶器描述于lepagemostefa等人的“apurificationrouteofbio-acrylicacidbymeltcrystallizationrespectfulofenvironmentalconstraints”,powdertechnology,vol.255,pp.98-102(2014)[“尊重环境的约束通过熔融结晶的生物丙烯酸的纯化路径”,粉末技术,第255卷,第98-102页(2014)]中,其中该丙烯酸在管的外表面上结晶。根据这些作者,这样的构型使得较大部分的初始熔体能够结晶而没有如果结晶是在管内的表面上发生的堵塞的风险,并且可以从该结晶器获得更高的生产率。诸位作者还要求保护来自他们的设计的其他益处,包括与先前已知的设计相比减少的循环时间。还有其他的结晶器设计继续引入该文献中,并且可以被考虑用于图2所示的熔融结晶阶段中的一个或多个,参见,例如由以下文献所描述的液压洗涤柱:verdoes和bassett“highpurityproductsbycrystallization”,specialtychemicals,vol.29,no.7,pp.32-35(2009)[“通过结晶的高纯度产物”,专用化学品,第29卷,第7期,第32-35页(2009)]和funakoshi等人“influencesofrefluxratioonseparationandpurificationofacrylicacidbyinclinedcolumncrystallizer”,journalofcrystalgrowth237-239,pp.2251-2256(2002)[“回流比对通过倾斜的柱结晶器的丙烯酸的分离和纯化的影响”,晶体生长杂志,237-239,第2251-2256页(2002)]。
降膜结晶总体上是在一个多管式交换器中进行,其中每个管在其顶部被连续进料一种有待从其中除去丙酸的丙烯酸的液体流(熔体),该液体沿该管的内壁下落为一个膜,在该管底部被接收并且在该管的顶部再循环持续只要在一个闭合回路中对于在该内部管壁上结晶所希望的量的丙烯酸是必要的。同时,一种热交换流体(典型地是乙二醇/水或甲醇/水)沿管的外壁流动并且提供结晶循环的每个阶段的操作所需的冷却或加热,其中从该管的底部到该管的顶部再循环持续结晶循环的持续时间。
每个结晶阶段本身在三相或三个阶段中进行:结晶,热析(sweating)和熔融。在该结晶阶段中,热交换流体的温度沿负温度梯度降低,从稍微高于熔体中的丙烯酸的结晶温度的温度开始,典型地为14摄氏度。晶体在该内部管壁的表面上形成。当大约30至80百分比的循环的丙烯酸已经结晶时,剩余的液体部分–母液–被排掉并且除去。在热析中,将热交换流体的温度沿着正温度梯度升高以便通过熔融除去以包含物的形式被捕获在正在形成的丙烯酸晶体的层中的杂质(在这种情况下,主要是丙酸);随着该层累积,这些包含物通过与随着丙烯酸结晶出来在丙酸中越来越富集的再循环不纯丙烯酸的接触渐增地出现。在该熔融阶段中,热交换液体的温度迅速增加到高于丙烯酸的熔点(14摄氏度)但不到由此发生聚合的程度(例如,不高于35至40摄氏度),并且该结晶层熔融并且被收集。典型地,将来自第一结晶器的结晶层供应到第二结晶器(作为熔体),使得通过顺序操作可以实现更高的纯度,如以下某些实例所示。
在图2中示意性示出的具体实施例中,该实施例利用如刚刚所述的色谱法和熔融结晶两者,第二闪蒸容器74闪蒸出在流76中的较轻组分,而剩余部分78由超过98%的纯丙烯酸组成,但仍含有超过优选的按重量计3000ppm上限的丙酸,然后被输送到第一熔融结晶阶段80。
来自阶段80中的母液82进入第二熔融结晶阶段84,而来自第二熔融结晶阶段84的结晶物86与来自第一熔融结晶阶段80的结晶物88组合,并且该组合的结晶物86和88被进料到第三熔融结晶阶段90中。将来自第二阶段84的母液92与来自在刚刚描述的典型的两-塔序列中的第二蒸馏塔的含丙酸的塔底物流94组合,并且这种组合用作至模拟移动床色谱法系统96的进料。在步骤40中来自优选的模拟移动床色谱系统的丙烯酸产物98然后连同结晶物86和88进料到第三熔融结晶阶段90,而来自模拟移动床色谱系统96的萃余液流100主要由粗丙烯酸产物50中所含的过量丙酸组成。
在一个实施例中,在任选的另外的步骤中,与丙酸一起保留在萃余液流100中的残余丙烯酸在反应器102中用氢气104氢化以产生另外的丙酸,并从而提供更高纯度的丙酸副产物106。在某些实施例中,该氢化可以以上面提及的授予dubois的us8,440,859中所描述的方式进行。然而,应该指出的是鉴于dubois考虑到被氢化的材料将含有按重量计从50至90百分比的丙烯酸,我们的萃余液100中的丙烯酸含量将是远远小于50重量百分比。因此,实现dubois的希望的至少85重量百分比、优选至少95重量百分比、和更优选至少99重量百分比的丙酸纯度应最终在我们的方法中显著更容易,其中,例如,萃余液100含有7.9重量百分比的剩余丙烯酸(实例28)而不是如dubois中主要由丙烯酸组成。
如在授予dubois的us8,440,859中涉及的,该氢化可以在液相或气相中用一种分子氢来源进行。由dubois提及的进行该氢化的已知方法包括fr2219927,chemickyprumsyl37,pp.651-653(1987)[化学工业37,第651-653页(1987)]和electroanalyticalchemistry(1975),pp.75-80[电分析化学(1975),第75-80页]。特别描述的是:使用一种钌-膦络合物和甲醇作为溶剂,在约60摄氏度下和约3mpa的压力下进行的均相液相方法;在250摄氏度与350摄氏度之间的温度下和0.1mpa与0.6mpa(从1至6个大气压)之间的压力下,在固定床中的氧化铝上的铜/锌催化剂上的多相气相催化;以及在从20至80摄氏度的温度和从0.1mpa至1.0mpa(1至10个大气压)的氢气压力下在钯催化剂上的多相催化,该钯催化剂以吸附在多孔载体(诸如硅酸或活性炭)上的液体钯盐溶液的形式施用,该盐随后被还原以便形成金属铂。
在另一个实施例中,通过较不优选的可替代的另外步骤,将在萃余液100中的过量丙酸氧化脱氢以提供额外的丙烯酸,例如,通过如在授予han等人的ep2039674b1中描述的一种催化剂和方法,其中使用具有式aambncxdzeof的混合金属氧化物催化剂,其中a是“至少一种选自由mo和w组成的组的元素;m是至少一种选自由v和ce组成的组的元素;n是至少一种选自由te、sb和se组成的组的元素;x是至少一种由以下项组成的元素:nb、ta、ti、al、zr、cr、mn、fe、ru、co、rh、ni、pt、sb、bi、b、in、as、ge、sn、li、na、k、rb、cs、fr、be、mg、ca、sr、ba、ra、hf、pb、p、pm、eu、gd、dy、ho、er、tm、yb和lu;并且z是至少一种选自下组的元素,该组由以下各项组成:zn、ga、ir、sm、pd、au、ag、cu、sc、y、pr、nd和tb;并且o是呈氧化物形式的氧并且其中,当a=1、b=0.01至1.0、c=0.01至1.0、d=0.01至1.0、e=0至0.1时,并且f是取决于其他元素的氧化状态”。优选的催化剂是“moavmtennbxoo和wavmtennbxoo,其中a、m、n、x和o[sic-f?]是如之前定义的”。可替代地,可以使用如在由han等人提及的jp2000053611参考文献中描述的mofecoo催化剂和方法。在另一个可替代的实施例中,如在授予keiko的jp07-330658(转让给大赛璐化学工业公司(daicelchemicalindustriesltd))中描述的一种催化剂和方法,其中使用一种具有式pamobvcadceebfog的催化剂使丙酸或其相应的酯氧化脱氢,其中a是以下项中的一种或多种:铜、砷、锑、硅、钨、铬、银和镁,b是以下以下项中的一种或多种:钾、铷、铯和铊,当(b)是12时,(a)是从0.5至3,(c)是从0.1至3,(d)是从0至3,(e)是从0.01至3,(f)是从0.01至2并且(g)是如所要求的。在另一个替代实施例中,可以如在mcentee等人,“羧酸的选择性催化氧化-脱氢-丙烯酸酯和巴豆酸酯在au/tio2界面上的形成(selectivecatalyticoxidative-dehydrogenationofcarboxylicacids–acrylateandcrotonateformationattheau/tio2interface)”,美国化学会志(j.am.chem.soc.),第136卷,第5116-5120页(2014)中描述的使用一种催化剂和方法,其中使用一种在二氧化钛上的金催化剂。在还另一个可替代的实施例中,可以如在授予watkins的us3,855,279中描述的使用一种催化剂和方法,其中(如具体示于实例9中)使用一种由铁和铅的混合磷酸盐的煅烧残留物组成的催化剂在氧的存在下并且在从250摄氏度至600摄氏度范围内的温度下可以将丙酸氧化脱氢为丙烯酸。这种另外的丙烯酸同样可以如本文所述的通过色谱法、通过结晶法或通过色谱法和结晶的组合进行加工。
冰丙烯酸产物流108(含有优选按重量计小于3000ppm的丙酸,更优选按重量计小于1000ppm的丙酸)是从第三熔融结晶阶段90产生,而将来自第三个熔融结晶阶段90的母液110再循环到结晶器序列的开始,到第一熔融结晶阶段80。
本发明通过以下非限制性的实例进一步进行说明。
实例
实例1
使用diaionamp-03两性离子交换树脂对丙烯酸/丙酸混合物进行一系列脉冲测试。标准测试程序涉及在室温下作为在水中的浆料将100ml的该树脂装入1.5cm直径的玻璃柱中。然后用500ml水洗涤该树脂。将水排到该树脂的顶部,然后将6ml的进料脉冲装入该树脂柱中。再次将该液体排到该树脂的顶部,并且添加2ml的水。再次,将该液体排到树脂的顶部,随后l0ml的水加入至顶部空间中。使水以3ml/分钟流动通过该树脂,同时每隔一段时间收集6ml馏分。然后分析这些6ml馏分。
在以上程序之后,如图3中所示发现乙酸和丙酸二者可以通过smb色谱使用一种两性离子交换树脂如diaionamp-03两性离子交换树脂在等度条件下与丙烯酸分离。
实例2-4
实例1中进行的脉冲测试显示:丙烯酸与丙酸的smb色谱分离是技术上可能的。然而,水要求将最有可能是相当显著的,由于后面的洗脱以及丙烯酸峰的轻微的拖尾。一种潜在的解决方案将是使用有机溶剂或者水与有机溶剂的混合物以降低洗脱要求。在以上程序之后,不同水平的甲醇和丙酮与水组合在实例2-4中进行评估以便看看是否丙烯酸的保留和峰形可以改善。
使用在水中5%丙酮(实例2和图4)表明丙烯酸峰的保留时间可以降低0.5床体积并且尾部降低约1床体积,表明相比于在smb色谱分离中的等度分离,洗脱要求实际上可降低。
在脉冲测试(实例3和图5)中在该洗脱中甲醇以15%作为共溶剂也降低了洗脱要求并且改进所有酸的峰的峰形。提高甲醇的相对浓度至50%(实例4和图6)显著地降低了该丙烯酸的洗脱时间但是丙烯酸和丙酸峰的峰重叠增加至其中该smb色谱分离将最可能不是成功的程度。
实例5-9
在实例1-4中报告的脉冲测试证实smb色谱可以使用等度条件和使用混合溶剂作为洗脱液二者用于丙烯酸与乙酸和丙酸二者的分离,尽管由于乙酸和丙烯酸的沸点差异,对于乙酸副产物,蒸馏分离可能是优选的。为了进一步评估这些不同的洗脱液在smb色谱安排中的性能,以2-5-4-1柱安排来安排12-柱转盘式(carousel)smb色谱单元,其采用diaionamp-03两性离子交换树脂(见图7)。独立地运行四个单独的泵用于解吸、富集、进料和再装流。
表1示出一系列使用该12-柱安排和等度条件的实验运行,其中报告的所有的流量是以克/分钟表示:
表1
如表1中的数据显示,实现了一种99+百分比纯的丙烯酸产物,相对于以大于95百分比的回收率的丙酸。进料包含从100-150g/升的丙烯酸结合有从7-15g/升的丙酸。
实例10-18
表2示出了一系列实验运行,使用该12-柱安排但使用在丙酮/水组合洗脱液中的10%丙酮:
表2
如从这些脉冲测试预期的,当将洗脱溶剂改变为包括10%丙酮时,所希望的产率和纯度以来自实例5-9的洗脱要求的显著降低实现:从用于等度分离的5:1洗脱:进料至用于混合的丙酮/水洗脱液的3:1。这导致增加的萃取物浓度和降低的蒸发。溶剂回收成本可能在一定程度上抵消这些益处。
实例19-27
表3示出用25%甲醇作为共溶剂进行的一系列实验运行:
表3
此外,以相比于等度操作的洗脱要求的显著降低,能实现所希望的产率和纯度两者。
实例28
将如图2示出的熔融结晶和色谱法序列通过以下方式建模:使用从马萨诸塞州柏林顿阿斯彭技术公司(aspentechnology,inc.,burlington,massachusetts)可商购的aspenplus(版本8.2)过程建模软件,遵循丙烯酸和丙酸的不同组合上进行的一系列的熔融结晶实验以便构建平衡相图并且确定丙烯酸与丙酸之间的共熔组成,并且进一步基于以上总结的色谱测试。该建模的结果于以下表4中示出,对于总体上根据授予paparizos等人的us4,786,756的用于制造生物基丙烯酸的先前方法中的来自第二蒸馏塔70的塔顶馏出物流72的进入剩余部分78和来自第二蒸馏塔70的含丙酸的塔底物流94。
表4
实例29-32
在来自发酵培养液的乳酸的萃取上进行了一系列批次试验,该发酵培养液是使用根据hara等人的us2012/0214214的方法生产并且然后通过超滤过滤。
将
然后将再生溶剂用于萃取另外量的超滤发酵培养液,并重复前述批次试验的步骤,直到四批超滤发酵培养液已经被处理。
四批次试验结果如下:
试验1:
超滤培养液体积500ml
培养液中的乳酸浓度71.6g/kg
溶剂混合物体积820ml
萃余液回收448ml
萃余液中的乳酸浓度10g/kg
萃余液的ph值未测定
添加的氢氧化铵28ml
回收的乳酸铵72ml
萃取的百分比乳酸87.2%
乳酸铵中的乳酸浓度34.8%
乳酸盐中的氨浓度8.32%
乳酸铵溶液浓度41.37%(计算的)
试验2
超滤培养液体积300ml
培养液中的乳酸浓度70.7g/kg
溶剂混合物体积800ml
萃余液回收276ml
萃余液中的乳酸浓度16.4g/kg
萃余液的ph值4.93
添加的氢氧化铵18ml
回收的乳酸铵52ml
萃取的百分比乳酸80%
乳酸铵中的乳酸浓度31.7%
乳酸盐中的氨浓度8.42%
乳酸铵溶液浓度37.7%(计算的)
试验3
超滤培养液体积300ml
培养液中的乳酸浓度70.7g/kg
溶剂混合物体积800ml
萃余液回收280ml
萃余液中的乳酸浓度12.64g/kg
萃余液的ph值4.77
添加的氢氧化铵17.5ml
回收的乳酸铵50ml
萃取的百分比乳酸83.4%
乳酸铵中的乳酸浓度33.4%
乳酸盐中的氨浓度9.39%
乳酸铵溶液浓度39.7%(计算的)
试验4
超滤培养液体积300ml
培养液中的乳酸浓度70.9g/kg
溶剂混合物体积810ml
萃余液回收277ml
萃余液中的乳酸浓度16.5g/kg
萃余液的ph值4.83
添加的氢氧化铵17.5ml
回收的乳酸铵48ml
萃取的百分比乳酸78.6%
乳酸铵中的乳酸浓度33.8%
乳酸盐中的氨浓度8.9%
乳酸铵溶液浓度40.2%(计算的)
实例33
对于本实施例和下一实施例,使用装备有疏水性x50聚丙烯管状膜(德国伍珀塔尔的迈博锐公司(membranagmbh,wuppertal,germany)的
在第一试验中,将1.25升超滤发酵培养液跨过在膜接触器中的x50管状膜萃取到2.5升的
实例34
在第二试验中,将1.14升超滤发酵培养液跨过在膜接触器中的x50管状膜萃取到2.5升的
- 还没有人留言评论。精彩留言会获得点赞!