一种新型稀土铕配合物及其制备方法与流程
本发明属于稀土配合物领域,具体涉及一种新型稀土铕配合物,特别地,涉及一种稀土铕配合物的制备方法。
背景技术:
稀土配合物具有发光强度高、有机物需要的激发能量小、荧光的效率高、能够很好地溶于有机溶剂等优势,因此,在越来越多的领域和方面被人们所应用。同时,在光纤通讯、荧光免疫、稀土分析抗肿瘤活性等一系列方面也具有潜在的应用价值。通过合成一种新型有机配体与稀土离子配位形成稀土配合物是有效提高稀土配合物发光效率的一种方法。一般有两种类型的有机配体与稀土离子可以形成配合物,一种是芳香羧酸类配体,一种是β-二酮类配体。申请号为201210528800.1的专利公开了一种稀土铕配合物,该配合物以β-二酮类配体(苯甲酰三氟丙酮)为第一配体、2-(2-苯并恶唑)为中性辅助配体。申请号为201410235707.0的专利公开了一种稀土铕配合物,该配合物以芳香型羧酸类大分子配体、邻菲罗啉为配体。其中,芳香羧酸类配体与β-二酮类配体各有优缺点,芳香羧酸类所形成的稀土配合物热稳定性高,但是不易升华;而β-二酮类配体易升华,但是热稳定性比较低。
技术实现要素:
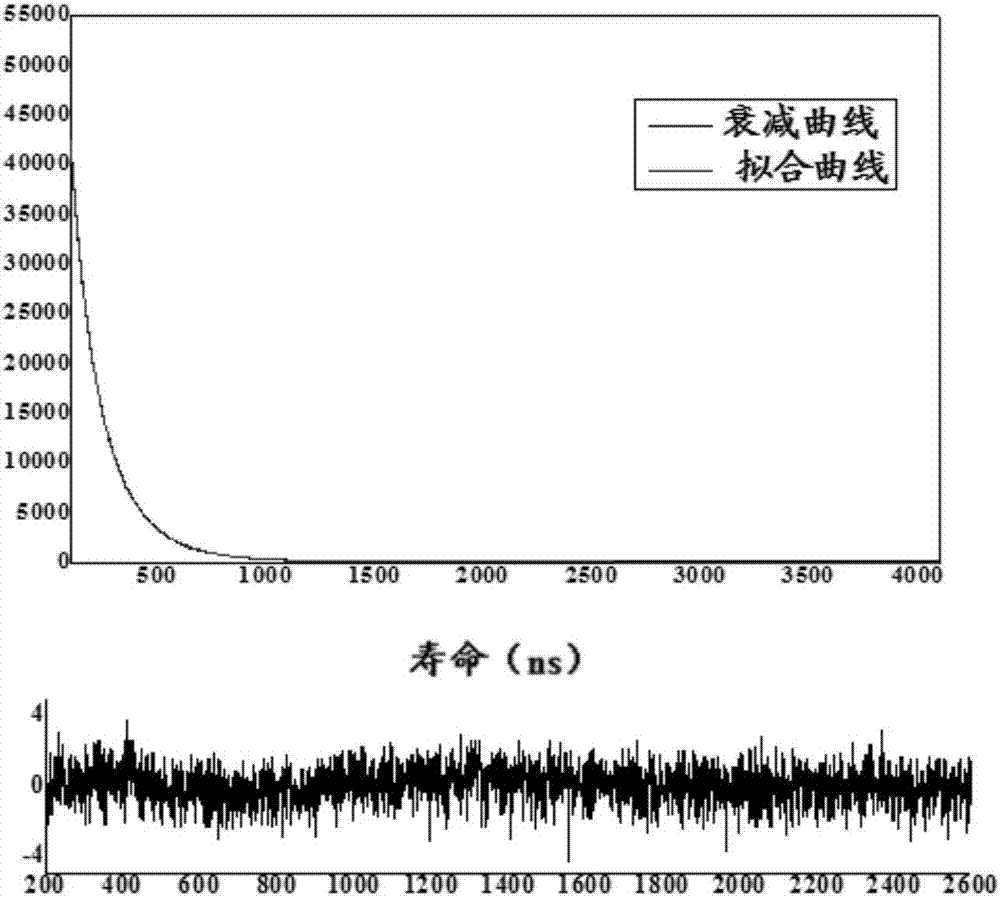
为了解决上述问题,本发明人进行了锐意研究,合成一种新型有机配体β-二酮类,使得在新配体上既存在芳香羧酸基团,又存在β-二酮基团,然后以该配体为第一配体、邻菲罗啉为第二配体制备一种新型稀土铕配合物,从而完成本发明。因此,本发明一方面提供一种新型稀土铕配合物的制备方法,该方法包括以下步骤:步骤1、合成β-二酮类配体:以苯乙酮与对苯二甲酸二甲酯为原料,进行酰基化反应,得到β-二酮类配体;步骤2、制备稀土铕配合物:以步骤1获得的β-二酮类配体作为第一配体,以邻菲罗啉作为第二配体,制备稀土铕配合物。其中,步骤1包括以下分步骤:步骤1.1、将对苯二甲酸二甲酯加入有机溶剂中,将甲醇钠加入甲醇中,然后将上述溶液混合得混合溶液;步骤1.2、将苯乙酮加入有机溶剂中,任选进行加热,优选沸腾后将其加入上述混合溶液中进行反应,优选反应结束后继续进行再反应,优选在冰水浴中进行,更优选在无水条件下进行;步骤1.3、对步骤1.2的产物依次调pH值、抽滤、取滤液、蒸馏,得到粗产品,将粗产品加入乙醇中,过滤、取固体、干燥,将得到的产物进行分离提纯,得到β-二酮类配体。步骤2包括以下分步骤:步骤2.1、将β-二酮类配体与EuCl3·6H2O加入乙醇,优选无水乙醇中,搅拌至溶解;步骤2.2、向步骤2.1的溶液中加入、优选滴加邻菲罗啉,优选加碱性介质调节pH值,搅拌,冷却过滤,得稀土铕配合物。本发明另一方面提供一种根据上述制备方法得到的稀土铕配合物,其中,所述稀土铕配合物为三元配合物,其中以三价稀土离子Eu3+为中心离子,β-二酮类配体为第一配体,邻菲罗啉为第二配体;所述β-二酮类配体由苯乙酮与对苯二甲酸二甲酯经酰基化反应制得,同时含有芳香羧基基团和β-二酮基团。本发明所具有的有益效果:(1)本发明所提供了一种稀土铕配合物的制备方法新颖,能够得到一种新型的稀土铕配合物;(2)本发明所提供的制备方法操作简单、容易实现;(3)本发明所提供的一种稀土铕配合物,其荧光寿命长、荧光强度高;(4)本发明所提供的一种稀土铕配合物,其包含一种新型的配体,在该配体上既存在芳香羧酸基团,又存在β-二酮基团,使得新配体易升华同时热稳定性好。附图说明图1示出根据实施例1制得的β-二酮类配体的在0~9ppm处的核磁共振氢谱图;图2示出根据实施例1制得的β-二酮类配体的在6.7~7.7ppm处的核磁共振氢谱图的放大图;图3示出根据实施例1制得的β-二酮类配体的在7.2~8.2ppm处的核磁共振氢谱图的放大图;图4示出根据实施例1制得的β-二酮类配体的在3.9~7.1ppm处的核磁共振氢谱图的放大图;图5示出根据实施例1制得的β-二酮类配体的质谱图;图6示出图5在a处的放大图;图7示出图5在b处的放大图;图8示出根据实施例1制得的β-二酮类配体的紫外光谱图;图9示出根据实施例1制得的β-二酮类配体的红外光谱图;图10示出邻菲罗啉的红外光谱图;图11示出根据实施例4制得的稀土铕三元配合物的红外光谱图;图12示出根据实施例1制得的β-二酮类配体的荧光发射谱图;图13示出根据实施例1制得的β-二酮类配体的荧光激发谱图;图14示出根据实施例4制得的稀土铕三元配合物与根据对比例1制得的稀土铕二元配合物的荧光发射光谱;图15示出根据实施例4制得的稀土铕三元配合物与根据对比例1制得的稀土铕二元配合物的荧光激发光谱;图16示出根据对比例1制得的稀土铕二元配合物的发射能级的衰减曲线及拟合曲线;图17示出根据实施例4制得的稀土铕三元配合物的发射能级的衰减曲线及拟合曲线。具体实施方式下面通过对本发明进行详细说明,本发明的特点和优点将随着这些说明而变得更为清楚、明确。根据本发明的一方面,提供了一种稀土铕配合物的制备方法,该方法包括以下步骤:步骤1、合成β-二酮类配体:以苯乙酮与对苯二甲酸二甲酯为原料,进行酰基化反应,得到β-二酮类配体;步骤2、制备稀土铕配合物:以步骤1获得的β-二酮类配体作为第一配体,以邻菲罗啉作为第二配体,制备稀土铕配合物。其中,所述步骤1的反应过程如下式进行,(1)为β-二酮类配体,(2)为副产物,所述β-二酮类配体又称对苯甲酰甲酸甲酯苯甲酰甲烷。步骤1包括以下分步骤:步骤1.1、将对苯二甲酸二甲酯加入有机溶剂中,将甲醇钠加入甲醇中,然后将上述溶液混合得混合溶液。其中,所述有机溶剂选自乙醚、乙醇、四氢呋喃、甲苯、正己烷中的一种或几种,优选为四氢呋喃;所述甲醇钠溶液的浓度为10%~50%,优选为20~40%,更优选为30%。步骤1.2、将苯乙酮加入有机溶剂中,任选地进行加热,优选沸腾后将其加入上述混合溶液中进行反应,优选反应结束后继续进行再反应,优选在冰水浴中进行,更优选在无水条件下进行。其中,所述有机溶剂选自乙醚、乙醇、四氢呋喃、甲苯、正己烷中的一种或几种,优选为四氢呋喃;有机溶剂的选择没有特别要求,只要能将苯乙酮溶解即可;所述加热于50~100℃进行,优选地,于70~80℃进行,更优选地,于75℃进行;加热的目的是为了让苯乙酮良好地溶解于有机溶剂中,因此,如果所选溶剂为苯乙酮的良溶剂,苯乙酮在其中在不加热的情况下也能良好溶解,不加热也可;但是,一般情况下,加热能够缩短苯乙酮的溶解时间以及提高其溶解效率,所以优选加热;所述反应如下进行:于无水条件下反应2~10小时,然后于40~90℃保温1~8个小时,优选的,于无水条件下反应4~8小时,然后于50~80℃保温2~6个小时,更优选地,于无水条件下反应6小时,然后于65℃保温4个小时;随着保温的进行,溶液由乳白色逐渐变为黄褐色,当保温结束后,溶液变为深黄褐色;其中,该反应的目标产物β-二酮类配体(1)(对苯甲酰甲酸甲酯苯甲酰甲烷)属于酯类,若反应环境中有水存在,会导致β-二酮类配体(1)水解而产生副产物(2),因此,该反应必须在无水条件下进行;由于反应温度接近或高于所用溶剂的沸点,因此,所述反应采用冷凝管进行回流,同时,为了实现无水条件,在冷凝管上面加干燥管,在干燥管内装有无水CaCl2。步骤1.3、对步骤1.2的产物依次调pH值、抽滤、取滤液、蒸馏,得到粗产品,将粗产品加入乙醇中,过滤、取固体、干燥,将得到的产物进行分离提纯,得到β-二酮类配体。其中,所述pH值为5~7,优选为6~7,在调节pH后,溶液由深黄褐色变为浅黄褐色;在一种优选的实施方式中,将粗产品加热溶于乙醇中,趁热过滤,重复4~5次,抽滤后干燥,优选为红外灯下干燥;所述粗产品是β-二酮类配体(1)与副产物(2)的混合物,为了得到高纯度的β-二酮类配体(1),需要对粗产物进行分离提纯;在一种优选的实施方式中,所述分离提纯采用柱色谱法;在进一步优选的实施方式中,选用石油醚与乙酸乙酯为淋洗液,更优选地,所述石油醚与乙酸乙酯以30:1配合;其中,副产物(2)的极性大于β-二酮类配体(1),因此,在柱分离过程中,首先被淋洗出的是副产物(2),后被淋洗出的是β-二酮类配体(1);在更进一步优选的实施方式中,在柱分离后,将β-二酮类配体(1)的淋洗液进行旋蒸得β-二酮类配体的纯产品。在一种优选的实施方式中,在步骤1中,所述对苯二甲酸二甲酯、甲醇钠、苯乙酮的摩尔比为1:(1~3):(0.2~1),优选为1:(1~2):(0.4~0.8),更优选为1:1:0.6。所述步骤2的反应过程如下式进行:步骤2包括以下分步骤:步骤2.1、将β-二酮类配体与EuCl3·6H2O加入乙醇,优选无水乙醇中,搅拌至溶解。其中,所述EuCl3·6H2O如下制得:将Eu2O3溶于稀盐酸中,缓慢蒸发析出结晶,抽滤,得EuCl3·6H2O。步骤2.2、向步骤2.1的溶液中加入、优选滴加邻菲罗啉,优选加碱性介质调节pH值,搅拌,冷却过滤,得稀土铕配合物。其中,所述碱性介质选自:氢氧化钠、氢氧化钾、氨水、三乙胺、吡啶中的一种或几种,优选为氨水、三乙胺、吡啶中的一种或几种,更优选为三乙胺;所述pH值为7~8。在一种优选的实施方式中,在步骤2中,所述β-二酮类配体、EuCl3·6H2O、邻菲罗啉的摩尔比为(2~5):(0.6~1.5):(0.6~1.5),优选为(2.5~3.5):(0.8~1.2):(0.8~1.2),更优选为3:1:1。本发明的另一方面提供一种由上述制备方法得到的稀土铕配合物,其分子式如下:如上式所示,所述稀土铕配合物为稀土铕三元配合物,其中以三价稀土铕离子Eu3+为中心离子,β-二酮类配体(对苯甲酰甲酸甲酯苯甲酰甲烷)为第一配体,邻菲罗啉为第二配体。所述稀土铕三元配合物具有较强的荧光强度,这是因为在邻菲罗啉作为第二配体引入后,其扩大了配合物的共轭π键范围,使稀土铕配合物的结构更加钢化,第二配体与铕(Ⅲ)能级相近,有利于能量的转移,使所述稀土铕三元配合物的荧光强度大大提高。所述稀土铕三元配合物的荧光寿命为2666.67ns,其拟合优度参数χ2为1.14。在稀土铕三元配合物的红外谱图中,所述稀土铕三元配合物在1726cm-1处有羰基的伸缩振动峰,在1649cm-1处有C=C-C(R)=O的伸缩振动峰,以及在1504cm-1处有-C=N-的特征伸缩吸收峰。在稀土铕三元配合物的荧光发射光谱中,在581nm、591nm以及613nm处出现了铕的特征发射峰。在一种优选的实施方式中,所述第一配体β-二酮类配体(对苯甲酰甲酸甲酯苯甲酰甲烷)由苯乙酮与对苯二甲酸二甲酯经酰基化反应制得,其中,所述β-二酮类配体同时含有芳香羧基基团和β-二酮基团。其中,芳香羧基基团有利于提高稀土铕三元配合物的热稳定性,β-二酮基团使该β-二酮类配体易于升华。实施例以下通过具体实例进一步描述本发明。不过这些实例仅仅是范例性的,并不对本发明的保护范围构成任何限制。实施例1β-二酮类配体的合成取对苯二甲酸二甲酯9.7g溶于100mLTHF中。将2.7g甲醇钠固体溶于6.4g的甲醇,配制成30%的甲醇钠溶液。将二者一起加入500mL单颈烧瓶中。将苯乙酮固体3.96g溶于25mLTHF中,油浴温度控制在75℃,待烧瓶中液体开始沸腾后将苯乙酮的溶液加入烧瓶中,该反应过程在无水条件下进行,用冷凝器进行回流,上面加干燥管,干燥管内装有无水CaCl2,反应时间为6小时。然后在65℃保温4个小时,可以看到溶液由原来的乳白色逐渐变为黄褐色。保温结束后,溶液变为深黄褐色。此时,将烧瓶从油浴锅中取下放置在5℃的冰水浴中,继续保持在无水条件下进行再反应。往烧瓶内滴加10%的盐酸0.5mL,溶液由深黄褐色变为浅黄色,pH为6-7之间。将烧瓶中溶液抽滤得到橙色的澄清溶液。蒸馏出溶剂后得浅黄褐色的粗产品。将所得粗产品加热溶于乙醇之中,趁热过滤。重复4-5次。抽滤得鹅黄色、粉末状的固体。置于红外干燥灯下干燥,再用石油醚与乙酸乙酯为30:1配成淋洗液进行柱色谱法提纯。最终可以得到装有纯产品的石油醚和乙酸乙酯混合液,经旋蒸仪可得纯产品β-二酮类配体。纯产品酯为白色,结晶状固体。实施例2β-二酮类配体的合成取对苯二甲酸二甲酯9.7g溶于100mLTHF中。将2.7g甲醇钠固体溶于10.8g的甲醇,配制成20%的甲醇钠溶液。将二者一起加入500mL单颈烧瓶中。将苯乙酮固体1.2g溶于15mLTHF中,油浴温度控制在70℃,待烧瓶中液体开始沸腾后将苯乙酮的溶液加入烧瓶中。该反应过程在无水条件下进行,用冷凝器进行回流,上面加干燥管,干燥管内装有无水CaCl2,反应时间为4小时。然后在50℃保温2个小时,可以看到溶液由原来的乳白色逐渐变为黄褐色。保温结束后,溶液变为深黄褐色。此时,将烧瓶从油浴锅中取下放置在2℃的冰水浴中,继续保持在无水条件下进行再反应。往烧瓶内滴加10%的盐酸0.5mL,溶液由深黄褐色变为浅黄色,pH为6-7之间。将烧瓶中溶液抽滤得到橙色的澄清溶液。蒸馏出溶剂后得浅黄褐色的粗产品。将所得粗产品加热溶于乙醇之中,趁热过滤。重复4-5次。抽滤得鹅黄色、粉末状的固体。置于红外干燥灯下干燥,再用石油醚与乙酸乙酯为30:1配成淋洗液进行柱色谱法提纯。最终可以得到装有纯产品的石油醚和乙酸乙酯混合液,经旋蒸仪可得纯产品β-二酮类配体。纯产品酯为白色,结晶状固体。实施例3β-二酮类配体的合成取对苯二甲酸二甲酯9.7g溶于100mLTHF中。将8.19g甲醇钠固体溶于12g的甲醇,配制成40%的甲醇钠溶液。将二者一起加入500mL单颈烧瓶中。将苯乙酮固体6g溶于45mLTHF中,油浴温度控制在80℃,待烧瓶中液体开始沸腾后将苯乙酮的溶液加入烧瓶中,该反应过程在无水条件下进行,用冷凝器进行回流,上面加干燥管,干燥管内装有无水CaCl2,反应时间为4小时。然后在80℃保温6个小时,可以看到溶液由原来的乳白色逐渐变为黄褐色。保温结束后,溶液变为深黄褐色。此时,将烧瓶从油浴锅中取下放置在8℃的冰水浴中,继续保持在无水条件下进行再反应。往烧瓶内滴加10%的盐酸0.5mL,溶液由深黄褐色变为浅黄色,pH为6-7之间。将烧瓶中溶液抽滤得到橙色的澄清溶液。蒸馏出溶剂后得浅黄褐色的粗产品。将所得粗产品加热溶于乙醇之中,趁热过滤。重复4-5次。抽滤得鹅黄色、粉末状的固体。置于红外干燥灯下干燥,再用石油醚与乙酸乙酯为30:1配成淋洗液进行柱色谱法提纯。最终可以得到装有纯产品的石油醚和乙酸乙酯混合液,经旋蒸仪可得纯产品β-二酮类配体。纯产品酯为白色,结晶状固体。实施例4稀土铕三元配合物的制备取3mmol的β-二酮类配体和1mmol的EuCl3·6H2O共同溶于10mL的无水乙醇中,微微加热,不断搅拌使β-二酮类配体完全溶解。然后取1mmol邻菲罗啉滴加到上述溶液中,然后再迅速取1.5mmol的三乙胺滴加到上述溶液中,可以看到有淡黄色沉淀生成,不断搅拌使反应完全。冷却后过滤,得淡黄色固体。实施例5稀土铕三元配合物的制备取2mmol的β-二酮类配体和0.6mmol的EuCl3·6H2O共同溶于10mL的无水乙醇中,微微加热,不断搅拌使β-二酮类配体完全溶解。然后取0.6mmol邻菲罗啉滴加到上述溶液中,然后再迅速取1mmol的三乙胺滴加到上述溶液中,可以看到有淡黄色沉淀生成,不断搅拌使反应完全。冷却后过滤,得淡黄色固体。实施例6稀土铕三元配合物的制备取5mmol的β-二酮类配体和1.5mmol的EuCl3·6H2O共同溶于15mL的无水乙醇中,微微加热,不断搅拌使β-二酮类配体完全溶解。然后取1.5mmol邻菲罗啉滴加到上述溶液中,然后再迅速取2mmol的三乙胺滴加到上述溶液中,可以看到有淡黄色沉淀生成,不断搅拌使反应完全。冷却后过滤,得淡黄色固体。对比例对比例1稀土铕二元配合物的制备取4mmol的β-二酮类配体和1mmol的EuCl3·6H2O共同溶于10mL的无水乙醇中,微微加热,不断搅拌使β-二酮类配体完全溶解。取1.5mmol的三乙胺滴加到上述溶液中,不断搅拌使反应完全,冷却后过滤。试验例试验例1熔点的测定取少许实施例1制得的β-二酮类配体装入到熔点测量管,高度约为3-5mm,开始检测。初熔温度为138.3℃,终熔温度为142.0℃。熔程为3.7℃。再重复一次该试验。初融温度为138.1℃,终熔温度142.1℃。熔程为4.0℃。根据上述实验结果可知β-二酮类配体的熔程小,即该样品的纯度较好。实施例2、实施例3制得的样品的熔点测试与实施例1相似。试验例2薄层色谱分析取少量实施例1制得的β-二酮类配体溶于乙醇中,待完全溶解之后,点板,放置到展开剂为石油醚与乙酸乙酯为15:1的混合溶液之中。待爬板结束,采用ZF5型紫外分析仪,在波长为254nm的紫外灯下仅有一个点,故而证明该样品纯度较好。实施例2、实施例3制得的样品的薄层色谱分析与实施例1相似。试验例3核磁分析采用德国布鲁克公司的300MHz的核磁共振波谱仪对实施例1制得的β-二酮类配体(对苯甲酰甲酸甲酯苯甲酰甲烷)进行核磁共振氢谱检测,如图1~4所示。其中,如图1~4所示,甲基上的氢出峰位置δ3.948(3H,d),亚甲基上的氢的出峰位置则为δ1.252(2H),是一个单峰。正常条件下,经过核磁拟合,亚甲基的出峰位置为δ3.8左右,但因为样品中混入了水再加之亚甲基上的的氢的活泼性很大,因此,尽管存在两个吸电子基团-羰基,氢同样可以向高场即向低化学位移方向移动。对于一取代的苯环上的4号碳原子上的氢则对应δ7.587(1H),δ7.584(1H),δ7.574(1H),是一个三重峰。对于该苯环上其他氢出峰位置分别为δ7.563(2H),δ7.550(2H),δ7.512(2H),δ7.492(2H),三重峰和δ8.144(2H),δ8.123(2H),双重峰。而对于第二个苯环上的氢所对应的出峰位置为δ8.034(2H),δ8.013(2H),双重峰和δ8.005(2H),δ7.987(2H),δ7.984(2H),三重峰。关于δ6.877的峰值是一个重水上的氢和之前活泼亚甲基上氢发生了移动所产生的峰二者发生了重叠。所以出现了一个较大的尖峰。而δ7.257则是氘代氯仿上氢的峰值。其他的峰是由于样品中少量杂质所引起的。从核磁谱图可以证明成功地合成了β-二酮类配体。实施例2、实施例3制得的样品的核磁谱图与与实施例1相似。试验例4质谱分析采用ICP-MS2000型电感耦合等离子质谱仪对实施例1制得的β-二酮类配体进行质谱分析,结果如图5所示,图6、图7分别为图5在a、b两处的放大图。其中,如图5~图7所示,该质谱图是对有机物样品β-二酮类配体加上一个钠原子处理后所测到的两个小图叠加而成,通过计算该有机物加上一个钠原子后的质荷比理论值为305。从图中可以看出,质荷比在305左右时相对强度最强,因此可以证明质荷比理论值和测量值是一致的。所以,加上一个钠原子后的相对分子量为305,分子式为C17H14O4Na。最终确定该有机物的相对分子量为282,即β-二酮类配体成功合成。实施例2、实施例3制得的样品的质谱谱图与与实施例1相似。试验例5紫外分析将实施例1制得的有机物样品β-二酮类配体溶于乙醇中,采用TU-1901型双光束紫外可见分光光度计对其进行紫外分析,得其紫外-可见吸收光谱,如图8所示。从图8中可以看出,该有机物在241nm处有一个较大的吸收峰,这是由于苯环结构的环状共轭共轭体系中π-π*电子跃迁,属于芳香族有机化合物的特征吸收峰,β二酮因为可以发生烯醇式互变,其π-π*跃迁也会对此造成影响。在285和297处也有两个较弱的吸收峰,这是因为碳氧双键的n→π*跃迁所造成的的,这种跃迁所需的能量较小,故可以在可见光区和近紫外区有较弱的吸收。再加之这两种跃迁会和苯环的伸缩振动发生重叠,故可以判断,该紫外光谱图对该有机物的光学性质的表征符合该有机物的结构。实施例2、实施例3制得的样品的紫外谱图与与实施例1相似。试验例6元素分析采用Perkin-Elmer公司的240B型自动元素分析仪对实施例1制得的β-二酮类配体与实施例4制得的稀土铕三元配合物分别进行元素分析检测,其检测结果如表1所示:表1:β-二酮类配体的元素分析结果由表1可以清楚地看到:对于β-二酮类配体,实际测得的碳含量与氢含量与理论值十分相近,其仅存的误差可能由于测试环境、测试仪器或测试人员的一些因素造成,由此可以断定,该产物为理论目标产物,其化学式如下:对于稀土铕三元配合物,实际测得的碳含量、氢含量以及氮含量与理论值十分相近,其仅存的误差可能由于测试环境、测试仪器或测试人员的一些因素造成,由此可以断定,该产物为理论目标产物,其化学式如下:实施例2、实施例3制得的样品的元素分析与与实施例1相似;实施例5、实施例6制得的样品的元素分析与与实施例4相似。试验例7红外分析采用WQF-510FTIR型红外光谱仪分别对邻菲罗啉、实施例1制得的β-二酮类配体以及实施例4制得的稀土铕配合物进行红外光谱测试,其中,采用压片法对实施例1制得的β-二酮类配体进行红外测试,如图9所示;用KBr压片法在4000-400cm-1波长之间分别对邻菲罗啉以及实施例4制得的稀土铕配合物进行红外测试,如图10~图11所示。如图9所示,为实施例1制得的β-二酮类配体的红外光谱图,其在3059cm-1和1604cm-1、1566cm-1、1437cm-1处的吸收峰说明有苯环的存在,其中在3059cm-1处的吸收峰为苯环上的C-H键伸缩振动所产生的吸收峰,后三个峰为苯环碳骨架伸缩振动的特征峰。在2922cm-1和2852cm-1处存在吸收峰,以及在1288cm-1处出现的峰,可以认为该样品中存在甲基,其中,在2922cm-1和2852cm-1处的吸收峰为烷基的伸缩振动。在1722cm-1处有一处很明显的吸收尖峰,这是羰基的特征峰,是碳氧双键的伸缩振动。而在2700cm-1处无吸收峰。所以可以判定该物质为酮类有机物。而在1288cm-1至1020cm-1出现的吸收峰则可以认为是Ar-H键的面内弯曲振动所产生的。在872cm-1到499cm-1之间出现的一系列强吸收峰则表明取代芳环的Ar-H键发生了面外弯曲振动。这些相关的峰验证了β-二酮类配体的成功合成。如图9~图11所示,分别为实施例1制得的β-二酮类配体、邻菲罗啉以及实施例4制得的稀土铕配合物的红外光谱。其中,与图10相比,图11在1726cm-1处有很强的羰基伸缩振动峰,在1408cm-1处出现了β-二酮类配体的相应特征峰,并且在1649cm-1处有C=C-C(R)=O的伸缩振动峰,这表明β-二酮类配体中的羰基氧参与了配位;在图11中,在3415cm-1出现了水的羟基伸缩振动峰,说明配合物中含有吸附水;其中,与图9相比,在图10中,邻菲罗啉-C=N-键的特征伸缩吸收峰(1560cm-1)在形成稀土配合物后向低波数移动1504cm-1,说明邻菲罗啉也参与了配位。实施例2、实施例3制得的样品的红外谱图与实施例1类似;实施例5、实施例6制得的样品的红外谱图与实施例4类似。试验例8荧光分析对实施例1制得的有机物样品β-二酮类配体做荧光测试得到其荧光发射谱图与荧光激发谱图,分别如图12、图13所示。其中,该有机物样品在紫外灯365nm的照射下能够发出淡绿色荧光,如图12所示,在440nm处为其最大发射波长,在此波长下,如图13所示,测得其最大激发波长为230nm,与试验例5中所得紫外-可见吸收光谱的最大吸收波长241nm相差不大。实施例2、实施例3制得的样品的荧光发射谱图以及荧光激发谱图与实施例1类似。对实施例4制得的稀土铕三元配合物以及对比例1制得的稀土铕二元配合物分别做荧光测试得到荧光发射光谱和荧光激发光谱,其中,稀土铕三元配合物与稀土铕二元配合物的荧光发射光谱如图14所示、稀土铕三元配合物与稀土铕二元配合物的荧光激发光谱如图15所示。其中,如图14所示,稀土铕三元配合物与稀土铕二元配合物的荧光发射光谱大致相同,均呈现三价铕离的特征发射,其中,581nm、591nm、613nm、653nm和698nm左右处的发射峰分别对应于Eu3+的5D0→7F0、5D0→7F1、5D0→7F2、5D0→7F3和5D0→7F4跃迁,其中以5D0→7F2最强,呈红色。如图15所示,稀土铕三元配合物与稀土铕二元配合物在280~365nm范围内有最大激发吸收峰,为π→π*的跃迁吸收,稀土铕三元配合物的特征荧光强度与稀土铕二元配合物相比较,其强度比值大于1,说明第二配体的加入对增强三价铕离子发光有明显的荧光增强协同作用。第二配体的这种荧光增强效应即称为第二配体的“协同效应”,其协同机理如下:由于第二配体扩大了共扼键的范围,起吸收能量和能量传递作用,稀土铕二元配合物一般都含有结晶水,而结晶水的O-H振子的振动能级可与稀土铕离子的电子能级偶合,产生非辐去活化用,而第二配体的加入可减少或消除结晶。实施例5、实施例6制得的样品的荧光发射谱图以及荧光激发谱图与实施例4类似。对对比例1制得的稀土铕二元配合物以及实施例4制得的稀土铕三元配合物分别进行荧光寿命分析。如图16所示,为对比例1制得的稀土铕二元配合物的发射能级的衰减曲线及拟合曲线,其中,稀土铕二元配合物经过三阶拟合得到其荧光寿命为666.67ns,拟合优度参数χ2为1.05,其中,衰减曲线与拟合曲线几乎重合;如图17所示,为实施例4制得的稀土铕三元配合物的发射能级的衰减曲线及拟合曲线,其中,稀土铕三元配合物经过二阶拟合得到其荧光寿命为2666.67ns,拟合优度参数χ2为1.14,其中,衰减曲线与拟合曲线几乎重合。实施例5、实施例6制得的样品的荧光荧光寿命以及拟合参数与实施例4类似。以上结合具体实施方式和范例性实例对本发明进行了详细说明,不过这些说明并不能理解为对本发明的限制。本领域技术人员理解,在不偏离本发明精神和范围的情况下,可以对本发明技术方案及其实施方式进行多种等价替换、修饰或改进,这些均落入本发明的范围内。本发明的保护范围以所附权利要求为准。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1