黄酮乙酸类衍生物、其药物组合物、其制备方法及用途与流程
技术领域
本发明属于医药化工领域,涉及一种黄酮乙酸类衍生物、其药物组合物、其制备方法及用途。
背景技术:
肿瘤需要依靠功能性的血管网络为其提供氧气、养料并及时清除代谢产物。除了可以通过与宿主血管整合而获得部分血管,肿瘤还必须通过形成新生血管网构建自己的血管系统才能持续地生长和发展。如果没有血管系统提供氧气和养料,实体瘤的增长不会超过1mm3。鉴于其在肿瘤发生发展中的重要作用,肿瘤血管已成为抗肿瘤治疗的一个重要靶点。
肿瘤血管破坏剂是可以快速并有选择性地破坏肿瘤血管的药物。它利用肿瘤血管和正常组织血管存在的差别选择性地破坏肿瘤血管,或借助能够特异结合肿瘤血管的配体将毒素、凝血诱导剂、凋亡诱导分子等运送到肿瘤血管,从而快速而有选择性地损坏或堵塞已构建完成的肿瘤血管。对于治疗血管已形成肿瘤,肿瘤血管破坏剂有显著的疗效。小分子肿瘤血管破坏剂可以快速,广泛地破坏已经形成的肿瘤血管,成为了研究的热点。目前已被普遍研究的肿瘤血管破坏剂包括秋水仙素、考布他汀A-4P、黄酮乙酸类等等,已应用于非小细胞肺癌、乳腺癌等实体瘤的治疗(Disrupting Tumour Blood Vessels,Nature Reviews,June 2005Volume 5:423-435)。
黄酮类肿瘤血管破坏剂是小分子肿瘤血管破坏剂的一种,目前普遍认为它是通过直接破坏肿瘤血管内皮细胞和促进肿瘤组织细胞释放肿瘤坏死因子α(TNF-α)等间接发挥破坏肿瘤血管的作用。黄酮乙酸(FAA)是最早被发现的黄酮类肿瘤血管破坏剂,它通过诱导肿瘤细胞产生TNF-α,显著升高肿瘤微环境中的TNF-α的浓度,使肿瘤局部的血管内皮细胞发生凋亡发挥作用,并且还能够避免TNF-α的全身毒性反应。但是FAA仅在小鼠肿瘤模型中有效,在临床试验中并没有抗癌作用。
5,6-二甲基呫吨酮-4-乙酸(DMXAA)是具有较高活性的FAA衍生物,活性强于FAA(研究发现FAA只在动物水平有效),但是研究表明其仅有轻微的剂量可逆毒性。此外,在III期临床试验中DMXAA未能显著延长患者的存活时间。
目前尚需要开发新的抗肿瘤化合物。
技术实现要素:
本发明人经过深入的研究和创造性的劳动,得到了一种新的黄酮乙酸类衍生物。本发明人惊奇地发现,该类化合物是有效的肿瘤血管破坏剂,具有较强的诱导肿瘤细胞产生TNF-α作用,以及对鸡胚绒毛尿囊膜当中血管的破坏作用,可以用于制备抗肿瘤药物。由此提供了下述发明:
本发明的一个方面涉及式I所示的化合物,或其可药用盐或酯,
其中:
R1选自H、C1-C6烷基;
当母环上的乙酸基团位于R1位置时,R1不存在;
R2选自苯基、一个或多个(例如2、3、4或5个)羟基取代的苯基,以及选自如下的基团:
具体地,对于R2为除苯环之外的上述其它取代基,R2与母环的连接位置为R2中的苯环上的未被取代的任意位点;
R3选自H、-OH。
本发明的另一方面涉及式II所示的化合物,或其可药用盐或酯,
其中:
R1选自H、C1-C6烷基;
当母环上的乙酸基团位于R1位置时,R1不存在;
R2选自苯基、一个或多个(例如2、3、4或5个)羟基取代的苯基,以及选自如下的基团:
具体地,对于R2为除苯环之外的上述其它取代基,R2与母环的连接位置为R2中的苯环上的未被取代的任意位点;
R3选自H、-OH;
表示单键或双键。
根据本发明任一项所述的式I或式II化合物,或其可药用盐,其中,
R1独立地选自H、C1-C3烷基;具体地,所述C1-C3烷基为甲基、乙基、丙基或异丙基;优选地,R1独立地为H或甲基。
根据本发明任一项所述的式I或式II化合物,或其可药用盐,其中,
R2独立地选自其中虚线表示R2与母环的连接位置。
根据本发明任一项所述的式I或式II化合物,或其可药用盐,其中,优选地,所述式I或式II化合物不包括以下4个化合物:
以及
根据本发明任一项所述的式I或式II化合物,其中,所述酯为母环上的乙酸基团形成的酯;具体地,为所述乙酸基团与C1-C6饱和一元醇(烷醇)形成的酯;具体地,所述C1-C6饱和一元醇为甲醇、乙醇、丙醇、异丙醇、正丁醇、戊醇或己醇。
本发明人还发现,本发明的酯亦是有效的肿瘤血管破坏剂,具有较强的诱导肿瘤细胞产生TNF-α作用,以及对鸡胚绒毛尿囊膜当中血管的破坏作用,可以用于制备抗肿瘤药物。
所述的式I或式II化合物,或其可药用盐或酯,其中,所述式I或式II化合物选自下面的表1:
表1:本发明的部分化合物
本发明的再一方面涉及本发明中任一项所述的式I化合物的制备方法,包括下述步骤:
(1)A片段的制备:
(2)B2、B3、B4、B5、B6片段的制备:
(3)以乙醇为溶剂,在60%氢氧化钾水溶液的作用下,将A片段分别与B1片段即B2片段、B3片段、B4片段、B5片段或B6片段反应得到式I化合物;
其中,虚线部分代表R1。
根据本发明任一项所述的制备方法,其中,所述步骤(1)包括:
a)以为原料与多聚甲醛和浓盐酸反应得到
b)在有机溶剂(例如甲苯)中,在试剂(例如四丁基溴化铵)存在下,使与氰化钠反应,得到的进一步水解,得到A片段(例如
根据本发明任一项所述的制备方法,其中,所述步骤(2)包括:
c)B1片段与B2片段可以直接购买;
d)B2片段与溴代异戊烯在碳酸钾溶液条件下反应,得到时B3片段
e)使B3片段与甲氧基甲基氯反应,得到B4片段
f)将B4片段分别与对甲苯磺酸、2,3-二氯-5,6-二氰基-对苯二醌,得到B5片段B6片段
根据本发明的详细教导以及已有的合成化学知识,本领域技术人员可以容易地合成本发明的式I化合物。
本发明的再一方面涉及本发明中任一项所述的式II化合物的制备方法,其包括本发明中任一项所述的制备式I化合物的方法,并且还包括:
(4)由式I化合物制得式II化合物;具体地包括下述步骤:
由式I化合物经DMSO/I2/微波条件下反应得到黄酮类化合物(例如参考实施例17);在TFA/微波条件下反应得到黄酮烷类化合物(例如参考实施例35);在双氧水/NaOH条件下反应得到式II化合物(例如参考实施例53)。
在一个实施方案中,本发明涉及制备通式I、式II所示的化合物的制备方法。具体地说,本发明提供了制备通式I、式II所示的化合物的方法,包括以下步骤:
1)A片段的合成
如反应路线,以邻羟基苯乙酮起始原料,将其与多聚甲醛和浓盐酸反应得到2-羟基-3-氯甲基-苯乙酮以及2-羟基-5-氯甲基-苯乙酮,将2-羟基-3-氯甲基-苯乙酮溶于甲苯中,在四丁基溴化铵存在下与氰化钠反应得到2-羟基-3-氰基-苯乙酮,将其与浓硫酸反应得到A1片段2-(3-乙酰基-2-羟苯基)乙酸,同样方法得到A2片段2-(3-乙酰基-4-羟苯基)乙酸。
以2-羟基-5-甲基-苯乙酮为原料,操作方法同上,得到A3片段2-(3-乙酰基-2-羟基-5-甲苯基)乙酸。
2)B片段的合成
如反应路线,B2片段与溴代异戊烯在碳酸钾水溶液中反应,得到B3片段;以丙酮为溶剂,B3片段与甲氧基甲基氯反应,得到B4片段;以甲苯为溶剂,B4片段分别与2,3-二氯-5,6-二氰基-对苯二醌、对甲苯磺酸反应,分别得到B5片段、B6片段。
c)以乙醇为溶剂,在60%氢氧化钾水溶液的作用下,A1片段、A2片段、A3片段分别与B1片段、B2片段、B3片段、B4片段、B5片段、B6片段反应得到式I化合物,式I化合物通过相应的反应得到式II化合物。
关于制备通式I、II化合物更详尽的资料还可见实施例。
本发明的再一方面涉及一种药物组合物,其包含本发明中任一项所述的式I或式II化合物或其可药用盐,以及任选的药学上可接受的辅料;可选地,其还包含脂多糖。
术语“组合物”意指包括包含指定量的各指定成分的产品,以及直接或间接从指定量的各指定成分的组合产生的任何产品。
通常本发明药物组合物含有0.1-90重量%的本发明化合物和/或其药学上可接受的盐。药物组合物可根据本领域已知的方法制备。用于此目的时,如果需要,可将本发明化合物和/或其药学上可接受的盐与一种或多种固体或液体药物赋形剂和/或辅剂结合,制成可作为人用的适当的施用形式或剂量形式。
本发明的化合物或含有它的药物组合物可以单位剂量形式给药,给药途径可为肠道或非肠道,如口服、肌肉、皮下、鼻腔、口腔粘膜、皮肤、腹膜或直肠等。给药剂型例如片剂、胶囊、滴丸、气雾剂、丸剂、粉剂、溶液剂、混悬剂、乳剂、颗粒剂、脂质体、透皮剂、口含片、栓剂、冻干粉针剂等。可以是普通制剂、缓释制剂、控释制剂及各种微粒给药系统。为了将单位给药剂型制成片剂,可以广泛使用本领域公知的各种载体。关于载体的例子是,例如稀释剂与吸收剂,如淀粉、糊精、硫酸钙、乳糖、甘露醇、蔗糖、氯化钠、葡萄糖、尿素、碳酸钙、白陶土、微晶纤维素、硅酸铝等;湿润剂与粘合剂,如水、甘油、聚乙二醇、乙醇、丙醇、淀粉浆、糊精、糖浆、蜂蜜、葡萄糖溶液、阿拉伯胶浆、明胶浆、羧甲基纤维素钠、紫胶、甲基纤维素、磷酸钾、聚乙烯吡咯烷酮等;崩解剂,例如干燥淀粉、海藻酸盐、琼脂粉、褐藻淀粉、碳酸氢钠与枸橼酸、碳酸钙、聚氧乙烯、山梨糖醇脂肪酸酯、十二烷基磺酸钠、甲基纤维素、乙基纤维素等;崩解抑制剂,例如蔗糖、三硬脂酸甘油酯、可可脂、氢化油等;吸收促进剂,例如季铵盐、十二烷基硫酸钠等;润滑剂,例如滑石粉、二氧化硅、玉米淀粉、硬脂酸盐、硼酸、液体石蜡、聚乙二醇等。还可以将片剂进一步制成包衣片,例如糖包衣片、薄膜包衣片、肠溶包衣片,或双层片和多层片。为了将给药单元制成丸剂,可以广泛使用本领域公知的各种载体。关于载体的例子是,例如稀释剂与吸收剂,如葡萄糖、乳糖、淀粉、可可脂、氢化植物油、聚乙烯吡咯烷酮、Gelucire、高岭土、滑石粉等;粘合剂如阿拉伯胶、黄蓍胶、明胶、乙醇、蜂蜜、液糖、米糊或面糊等;崩解剂,如琼脂粉、干燥淀粉、海藻酸盐、十二烷基磺酸钠、甲基纤维素、乙基纤维素等。为了将给药单元制成栓剂,可以广泛使用本领域公知的各种载体。关于载体的例子是,例如聚乙二醇、卵磷脂、可可脂、高级醇、高级醇的酯、明胶、半合成甘油酯等。为了将给药单元制成胶囊,将有效成分化合物或其可药用盐与上述的各种载体混合,并将由此得到的混合物置于硬的明明胶囊或软胶囊中。也可将有效成分化合物或其可药用盐制成微囊剂,混悬于水性介质中形成混悬剂,亦可装入硬胶囊中或制成注射剂应用。为了将给药单元制成注射用制剂,如溶液剂、乳剂、冻干粉针剂和混悬剂,可以使用本领域常用的所有稀释剂,例如,水、乙醇、聚乙二醇、1,3-丙二醇、乙氧基化的异硬脂醇、多氧化的异硬脂醇、聚氧乙烯山梨醇脂肪酸酯等。另外,为了制备等渗注射液,可以向注射用制剂中添加适量的氯化钠、葡萄糖或甘油,此外,还可以添加常规的助溶剂、缓冲剂、pH调节剂等。
此外,如需要,也可以向药物制剂中添加着色剂、防腐剂、香料、矫味剂、甜味剂或其它材料。
本发明的化合物,或其可药用盐的给药剂量取决于许多因素,例如所要预防或治疗疾病的性质和严重程度,患者或动物的性别、年龄、体重及个体反应,所用的具体化合物,给药途径及给药次数等。上述剂量可以单一剂量形式或分成几个,例如二、三或四个剂量形式给药。
可改变本发明药物组合物中各活性成分的实际剂量水平,以便所得的活性化合物量能有效针对具体患者、组合物和给药方式得到所需的治疗反应。剂量水平须根据具体化合物的活性、给药途径、所治疗病况的严重程度以及待治疗患者的病况和既往病史来选定。但是,本领域的做法是,化合物的剂量从低于为得到所需治疗效果而要求的水平开始,逐渐增加剂量,直到得到所需的效果。
本发明的再一方面涉及中任一项所述的式I或式II化合物或其可药用盐或者本发明的药物组合物在制备如下任一项所述的药物中的用途:
1)调节例如上调肿瘤坏死因子α水平的药物;
2)诱导肿瘤细胞产生肿瘤坏死因子α的药物‘
3)破坏肿瘤血管或者抑制肿瘤血管形成的药物;
4)抑制肿瘤细胞或者血管内皮细胞的药物;以及
5)治疗和/或预防和/或辅助治疗肿瘤的药物中的用途;
具体地,所述肿瘤细胞为结肠癌细胞(例如DLD-1人结肠癌细胞)、RAW 264.7巨噬细胞、乳腺癌细胞(例如MDA-MB-231乳腺癌细胞)或宫颈癌细胞(例如Hela宫颈癌细胞)。
具体地,所述肿瘤为肺癌、乳腺癌、宫颈癌或结肠癌。
本发明的再一方面涉及一种选自如下任一项所述的方法,包括使用有效量的本发明中任一项所述的式I或式II化合物或其可药用盐或者本发明的药物组合物的步骤:
1)在体内或体外调节例如上调肿瘤坏死因子α水平的方法;
2)在体内或体外诱导肿瘤细胞产生肿瘤坏死因子α的方法;以及
3)在体内或体外抑制肿瘤细胞例如抑制肿瘤细胞增殖和/或迁移的方法。
在本发明的一个实施方案中,上述的在体外的方法是非治疗目的的。
本发明的再一方面涉及一种治疗和/或预防和/或辅助治疗肿瘤的方法,包括使用有效量的本发明中任一项所述的式I或式II化合物或其可药用盐或者本发明的药物组合物的步骤;具体地,所述肿瘤为肺癌、乳腺癌、宫颈癌、结肠癌。
术语“有效量”是指可在受试者中实现治疗、预防、减轻和/或缓解本发明所述疾病或病症的剂量。
术语“受试者”可以指患者或者其它接受本发明组合物以治疗、预防、减轻和/或缓解本发明所述疾病或病症的动物,特别是哺乳动物,例如人、狗、猴、牛、马等。
术语“疾病和/或病症”是指所述受试者的一种身体状态,该身体状态与本发明所述疾病和/或病症有关。
但应认识到,本发明化合物或药物组合物的总日用量须由主诊医师在可靠的医学判断范围内作出决定。对于任何具体的患者,具体的治疗有效剂量水平须根据多种因素而定,所述因素包括所治疗的障碍和该障碍的严重程度;所采用的具体化合物的活性;所采用的具体组合物;患者的年龄、体重、一般健康状况、性别和饮食;所采用的具体化合物的给药时间、给药途径和排泄率;治疗持续时间;与所采用的具体化合物组合使用或同时使用的药物;及医疗领域公知的类似因素。例如,本领域的做法是,化合物的剂量从低于为得到所需治疗效果而要求的水平开始,逐渐增加剂量,直到得到所需的效果。一般说来,本发明的化合物用于哺乳动物特别是人的剂量可以介于0.001-1000mg/kg体重/天,例如介于0.01-100mg/kg体重/天,例如介于0.01-10mg/kg体重/天。
根据本发明的化合物可以有效地预防和/或治疗本发明所述的各种疾病或病症。
本发明的再一方面涉及选自如下中间体化合物:
以及
本发明的再一方面涉及本发明中所述的任一中间体化合物在制备抗肿瘤药物或者在制备本发明任一项所述的式I或式II化合物中的用途。
发明的有益效果
本发明的化合物仅有较强的诱导肿瘤细胞产生TNF-α的作用,以及对鸡胚绒毛尿囊膜当中血管的破坏作用,是有效的肿瘤血管抑制剂或者破坏剂,具有用于制备抗肿瘤药物的前景。此外,本发明的化合物不依赖LPS途径也具有TNF-α促分泌作用。
附图说明
图1:control(对照)、DMXAA、实施例2化合物的瘤体图片。上方刻度尺可读出瘤体大小。
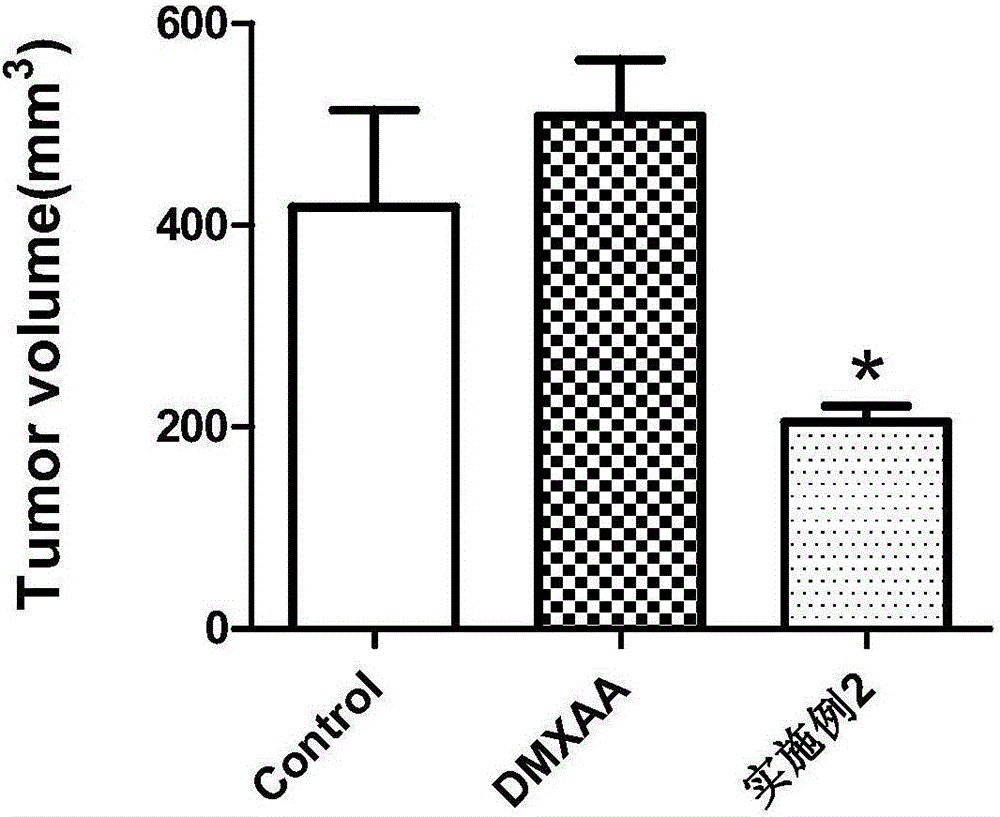
图2:瘤体体积变化。*P<0.05.n=3-5只鼠/组。mean±SEM。
图3:对血管破坏作用(Hoshest 33342染色)。图3A,control(对照);图3B,DMXAA;图3C,实施例2化合物。
具体实施方式
下面将结合实施例对本发明的实施方案进行详细描述,但是本领域技术人员将会理解,下列实施例仅用于说明本发明,而不应视为限定本发明的范围。实施例中未注明具体条件者,按照常规条件或制造商建议的条件进行。所用试剂或仪器未注明生产厂商者,均为可以通过市购获得的常规产品。
化合物的熔点由RY-1熔点仪测定,温度计未较正。质谱由Micromass ZabSpec高分辨率质谱仪(分辨率1000)测定。1H-NMR由JNM-ECA-400超导NMR仪测定,工作频率1H-NMR 400MHz。
在本发明的上下文中,使用如下缩略语:
MOM:-甲氧基甲基
DMSO:二甲基亚砜
DDQ:2,3-二氯-5,6-二氰基-对苯二醌
TsOH:对甲苯磺酸
下面通过制备例1-7来制备中间体。
制备例1:2-羟基-3-氯甲基-苯乙酮的制备
反应流程:
步骤1:邻羟基苯乙酮衍生物的制备
机械搅拌下,邻羟基苯乙酮178g(1.30mol)、多聚甲醛39.5g(1.30mol)与浓盐酸800mL在50-60℃下反应7小时。分出有机相、水相,以甲苯200mL提取水相,用甲苯600mL溶解反应体系下层油状物,合并有机相,有机相依次用饱和NaHCO3水溶液300mL×2次,饱和食盐水300mL洗涤,无水硫酸钠干燥。过滤、旋转蒸发仪蒸出溶剂后,对残余油状物进行减压蒸馏,得到主馏分有211.4g,是2-羟基-3-氯甲基-苯乙酮和2-羟基-5-氯甲基-苯乙酮的混合物。按照1g~5mL的比例向其中加入正己烷,加热溶解。冷却后析出晶体为2-羟基-5-氯甲基-苯乙酮155.1g;过滤后滤液冷藏(4℃)即有油状物析出,倾出上清液,浓缩约一半后放置即可析出大量晶体,此晶体为2-羟基-3-氯甲基-苯乙酮38.3g。总收率80.8%。
制备例2:2-羟基-5-氰基甲基-苯乙酮的制备
反应流程:
2-羟基-5-氯甲基-苯乙酮37g(0.2mol)溶解在500mL甲苯当中(加热促溶),加入四丁基溴化铵3.2g(20mmol),室温搅拌下,向其中滴加氰化钠12.5g(0.255mol)溶解在80mL水当中的溶液。20分钟滴完,70℃反应1小时。
冷却,有机相以500mL×2水洗涤,有机相干燥,过滤,减压浓缩得到大量固体。用石油醚/乙酸乙酯(5:1)重结晶,得到纯品31.6g,收率89.2%。
2-羟基-3-氰基甲基-苯乙酮的制备参照上述操作。
制备例3:2-(3-乙酰基-4-羟苯基)乙酸的制备
反应流程:
2-羟基-5-氰基-苯乙酮8.7g(34.5mmol),加入33%的硫酸75mL(1:2体积比稀释得到),回流反应1.5小时。冷却,加入冷水100mL,放置,析出大量黄色固体。过滤水洗,粗品用热水重结晶,热滤去除杂质,滤液析出大量淡黄色固体,过滤得到纯产物8.3g。收率87.7%。
2-(3-乙酰基-2-羟苯基)乙酸的制备参照上述操作。
制备例4:4-羟基-3-(3-亚甲基-2-烯)苯甲醛的制备
反应流程:
碳酸钾140g(1mol)溶解于200g水中,待放热结束后,加入对羟基苯醛61.0g(0.5mol),搅拌至完全溶解。再滴加溴代异戊烯60.0mL(0.51mol),5分钟内滴完。室温下反应,TLC检测12小时。
反应液以乙醚250mL×2萃取,水相弃去;乙醚层合并,以5%碳酸钠水溶液200mL×3洗涤,水相弃去;再用0.5N氢氧化钠水溶液200mL×3洗涤,有机相弃去;水相用浓盐酸酸化后,以乙醚200mL×3萃取,干燥,层析柱分离,石油醚/乙酸乙酯(5:1),得到14.1g无色油状物,放置缓慢固化。收率14.8%。
制备例5:4-(甲氧基甲氧基)-3-(3-亚甲基-2-烯)苯甲醛的制备
反应流程:
4-羟基-3-(3-亚甲基-2-烯)苯甲醛1.9g(10mmol),溶解在50mL丙酮当中,加入碳酸钾2.0g,冰浴搅拌下加入甲氧基甲基氯2.0mL(20mmol),10分钟滴完,然后撤去冰浴,在常温下反应8小时。减压旋干溶剂,加入乙醚30mL,分别以水20mL、0.5N氢氧化钠溶液20mL、饱和食盐水20mL洗涤,干燥,过滤,层析柱分离,石油醚/乙酸乙酯(8:1),得无色油状物2.1g,放置逐渐固化,收率83.1%。
制备例6:2,2-二甲基-2H-苯并吡喃-6-甲醛的制备
反应流程:
4-(甲氧基甲氧基)-3-(3-亚甲基-2-烯)苯甲醛1.90g(10mmol)溶解在30mL甲苯当中,加入DDQ(2,3-二氯-5,6-二氰基-对苯二醌)2.70g(12mmol),加热回流5小时。冷却后过滤,滤饼用甲苯10mL洗涤,滤液依次用15mL水、饱和碳酸钾溶液、饱和氯化钠溶液洗涤。有机相干燥,过滤,层析柱分离,石油醚/乙酸乙酯(10:1),得到产物0.80g,收率44.4%。
制备例7:2,2-二甲苯并吡喃-6-甲醛的制备
反应流程:
4-(甲氧基甲氧基)-3-(3-亚甲基-2-烯)苯甲醛1.90g(10mmol)溶解在甲苯30mL当中,加入对甲苯磺酸0.20g,回流3小时。冷却后过滤,滤饼用甲苯10mL洗涤,滤液依次用15mL水、饱和碳酸钾溶液、饱和氯化钠溶液洗涤。有机相干燥,过滤,层析柱分离,石油醚/乙酸乙酯(10:1),得到产物1.58g,收率83.2%。
实施例2:2-(2-羟基-3-(3-(4-羟基苯基)丙烯酰基)苯基)乙酸
将2-(3-乙酰基-2-羟苯基)乙酸1.00g(5.18mmol)和对羟基苯醛0.63g(5.18mmol)混合,加入乙醇6mL溶解,加入60%氢氧化钾水溶液8mL,室温反应4小时。用冰水50mL稀释,用乙醚20mL萃取,乙醚相弃去。冰浴搅拌下,水相用盐酸酸化至pH≈2,有大量明黄色固体析出,过滤干燥,得0.89g产物,收率92.2%。
1H-NMR(DMSO-d6,400MHz):3.586(2H,s,-CH2-COOH),6.843-6.981(3H,m,Ar-H×3),7.498-7.517(1H,dd,Ar-H),7.806-7.893(4H,m,Ar-H×4),8.253-8.273(1H,m,Ar-H),10.232(1H,s,-OH),12.292(1H,s,-OH),13.492(1H,s,-COOH)。ESI-MS[M+1]:299.1。
实施例3:2-(4-羟基-3-(3-(4-(羟基苯基)丙烯酰基)苯基)乙酸
参照实施例2的合成方法制备本实施例化合物,不同的是使用2-(3-乙酰基-4-羟苯基)乙酸1.00g(5.18mmol)代替相应起始原料,得到产品1.45g,收率94.1%。
1H-NMR(DMSO-d6,400MHz):3.603(2H,s,-CH2-COOH),6.778-7.018(3H,m,Ar-H×3),7.326-7.465(2H,m,Ar-H×2),7.658-7.823(3H,m,Ar-H×3),8.143-8.148(1H,d,J=2Hz,Ar-H),10.231(1H,s,-OH),12.367(1H,s,-OH),12.729(1H,s,-COOH)。ESI-MS[M+1]:298.0。
实施例4:2-(2-羟基-3-(3-(4-羟基苯基)丙烯酰基)-5-甲基苯基)乙酸
参照实施例2的合成方法制备本实施例化合物,不同的是2-(3-乙酰基-2-羟基-5-甲苯基)乙酸1.00g(4.83mmol)代替相应起始原料,得到产品1.43g,收率88.5%。
1H-NMR(DMSO-d6,400MHz):2.325(3H,s,-CH3),3.552(2H,s,-CH2-COOH),6.855-6.877(2H,d,J=8.8Hz,Ar-H×2),7.336-7.340(1H,d,J=1.6Hz,Ar-H),7.818-7.934(4H,m,Ar-H×4),8.093-8.096(1H,d,J=1.2Hz,Ar-H),10.235(1H,s,-OH),12.280(1H,s,-OH),13.310(1H,s,-COOH)。ESI-MS[M+1]:313.0。
实施例9:2-(3-(3-(2,2-二甲基-6-基)丙烯酰基)-2-羟基苯基)乙酸
参照实施例2的合成方法制备本实施例化合物,不同的是使用2-(3-乙酰基-4-羟苯基)乙酸1.65g(8.55mmol)和4-(甲氧基甲氧基)-3-(3-亚甲基-2-烯)苯甲醛2.00g(8.55mmol)代替相应起始原料,得到产品1.30g,收率37.14%。
1H-NMR(DMSO-d6,400MHz):1.688-1.730(6H,d,J=16.8Hz,-CH3×2),3.323(1H,s,-CH2之一),3.391(3H,s,-CH3),3.618(2H,s,-CH2-COOH),5.276-5.317(3H,m,-OCH2;-CH),6.943-6.964(1H,d,J=8.4Hz,Ar-H),7.111-7.133(1H,d,J=8.8Hz,Ar-H),7.444-7.471(1H,dd,Ar-H),7.713-7.775(2H,m,Ar-H×2),7.814-7.893(2H,m,Ar-H×2),8.110-8.115(1H,d,J=2Hz,Ar-H),12.591(2H,s,-OH;-COOH)。ESI-MS[M+1]:411.5。
实施例10:2-(2-(2-羟基-3-(3-(4-(甲氧基甲氧基)-3-(3-甲基丁-2-烯基)苯基)丙烯酰基)-5-甲基苯基)乙酸
参照实施例2的合成方法制备本实施例化合物,不同的是使用2-(3-乙酰基-2-羟基-5-甲苯基)乙酸1.77g(8.55mmol)和4-(甲氧基甲氧基)-3-(3-亚甲基-2-烯)苯甲醛2.00g(8.55mmol)代替相应起始原料,得到产品1.71g,收率47.2%。
1H-NMR(DMSO-d6,400MHz):1.695-1.741(6H,d,J=18.4,-CH3×2),2.341(3H,s,-CH3),3.334-3.415(5H,m,-CH2-CH;-OCH3),3.562(2H,s,-CH2-COOH),5.287-5.326(3H,m,-OCH2;-CH2-CH),7.116-7.139(1H,d,J=9.2Hz,Ar-H),7.357-7.360(1H,d,J=1.2Hz,Ar-H),7.776-7.859(3H,m,Ar-H×3),7.943-7.982(1H,d,J=15.6Hz,Ar-H),8.089(1H,s,Ar-H),12.296(1H,s,-OH),13.245(1H,s,-COOH)。ESI-MS[M+1]:425.2。
实施例1、5、6、7、8、11、12、13、14、15、16、68的合成参照实施例2,如下面的表2。
表2:参照实施例2合成的部分化合物
实施例17:2-(2-(4-羟基苯基)-4-氧代-4H-苯并吡喃-8-基)乙酸
将2-(2-羟基-3-(3-(4-羟基苯基)丙烯酰基)苯基)乙酸89.4mg(0.3mmol),溶于2mL DMSO当中,加入3mg I2,微波反应10分钟,设定温度150℃。用乙酸乙酯15mL和水15mL萃取,有机相干燥,过滤,浓缩,层析柱分离,二氯甲烷/甲醇(20:1),得到黄色固体67mg,收率54.4%。
1H-NMR(DMSO-d6,400MHz):3.987(2H,s,-CH2-COOH),6.859-6.943(3H,m,Ar-H×3),7.400-7.438(1H,t,J=7.6Hz,Ar-H),7.714-7.732(1H,dd,Ar-H),7.920-7.961(3H,m,Ar-H×3),10.284(1H,s,-OH),12.544(1H,s,-COOH).ESI-MS[M+1]:297.1。
实施例18:2-(3-羟基-2-(4-羟基苯基)-4-氧代-4H-苯并吡喃-8-基)乙酸
参照实施例17的合成方法制备本实施例化合物,不同的是使用2-(4-羟基-3-(3-(4-羟基苯基)丙烯酰基)苯基)乙酸0.3g(1.01mmol)
为起始原料,得到产品0.16mg,收率60.4%。
1H-NMR(DMSO-d6,400MHz):3.757(2H,s,-CH2-COOH),6.862-6.955(3H,m,Ar-H×3),7.699-7.701(2H,d,J=0.8Hz,Ar-H×2),7.922-7.975(3H,m,Ar-H×3),10.318(1H,s,-OH),12.444(1H,s,-COOH).ESI-MS[M+1]:297.1。
实施例20:2-(2-(4-羟基-3-(3-甲基丁-2-烯基)苯基)-4-氧代-4H-苯并吡喃-8-基)乙酸
参照实施例17的合成方法制备本实施例化合物,不同的是使用2-(2-羟基-3-(3-(4-羟基-3-(3-甲基丁-2-烯基)苯基)丙烯酰基)苯基)乙酸0.25g(0.68mmol)为起始原料,得到产品135mg,收率54.4%。
1H-NMR(DMSO-d6,400MHz):1.719(6H,s,-CH3×2),3.981(2H,s,-CH2-COOH),5.340-5.378(1H,t,J=7.6Hz,-CH2-CH),6.851-6.951(2H,m,Ar-H×2),7.402-7.439(1H,t,J=7.2Hz,Ar-H),7.711-7.804(3H,m,Ar-H×3),7.933-7.956(1H,dd,Ar-H),10.266(1H,s,-OH),12.585(1H,s,-COOH).ESI-MS[M+1]:365.2。
实施例22:2-(2-(4-羟基-3-(3-甲基丁-2-烯基)苯基)-6-甲基-4-氧代-4H-苯并吡喃-8-基)乙酸
参照实施例17的合成方法制备本实施例化合物,不同的是使用2-(2-羟基-3-(3-(4-羟基-3-(3-甲基丁-2-烯基)苯基)丙烯酰基)-5-甲基苯基)乙酸0.26g(0.68mmol)为起始原料,得到产品134mg,收率51.8%。
1H-NMR(DMSO-d6,400MHz):1.715(6H,S,-CH3×2),2.410(3H,S,Ar-CH3),3.335(2H,S,-CH2-CH),3.929(2H,S,-CH2-COOH),5.350-5.356(1H,t,J=1.2Hz,-CH2-CH),6.808-6.943(2H,m,Ar-H×2),7.538-7.543(1H,d,J=2Hz,Ar-H),7.737-7.779(3H,m,Ar-H×3),10.245(1H,s,-OH),12.580(1H,s,-COOH).ESI-MS[M+1]:379.1。
实施例26:2-(2',2'-二甲基-4-氧代-2'H,4H-2,6'-苯并吡喃-8-基)乙酸
参照实施例17的合成方法制备本实施例化合物,不同的是使用2-(3-(3-(2,2-二甲基-2H-苯并吡喃-6-基)-2-丙烯酰基)-2-羟基苯)乙酸110mg(0.3mmol)为起始原料,得到产品54.4mg,收率49.8%。
1H-NMR(DMSO-d6,400MHz):1.318-2.222(6H,m,-CH3×2),2.852-2.918(1H,m,-CH2之一),3.056-3.091(1H,m,-CH2之一),3.230-3.319(3H,m,-CH;-CH2),4.003(2H,s,-CH2-COOH),6.891-6.927(2H,d,J=5.2Hz,Ar-H×2),7.431-7.450(1H,t,J=7.6Hz,Ar-H),7.892-7.961(4H,m,Ar-H×4),12.646(1H,s,-COOH).ESI-MS[M+1]:365.1。
实施例19、21、23、24、25、27、28、29、30、31的合成参照实施例17,如下面的表3。
表3:参照实施例17合成的部分化合物
实施例35:2-(2-(4-羟基苯基)-4-氧代苯并二氢吡喃-8-基)乙酸
将2-(2-羟基-3-(3-4-羟基苯基)丙烯酰基)苯基)乙酸0.25g(0.84mmol),溶于2mL三氟乙酸当中,微波反应10分钟,设定温度80℃。用乙酸乙酯20mL稀释后,用水15mL×2洗涤,有机相干燥,过滤浓缩,层析柱分离,二氯甲烷/甲醇(35:1),得到黄色固体0.11g,收率44%。
1H-NMR(DMSO-d6,400MHz):2.796-2.846(1H,dd,-CH2之一),3.120-3.195(1H,m,-CH2之一),3.580(1H,s,-CH2-COOH),5.482-5.521(1H,s,-CH),6.772-6.801(2H,m,Ar-H×2),7.016-7.054(1H,t,J=7.6Hz,Ar-H),7.305-7.327(2H,d,J=8.6Hz,Ar-H),7.506-7.529(1H,dd,Ar-H),7.690-7.715(1H,dd,Ar-H),9.579(1H,s,-OH),12.335(1H,s,-COOH)ESI-MS[M+1]:299.1。
实施例38:2-(2-(4-羟基-3-(3-甲基丁-2-烯基)苯基)-4-氧代苯并二氢吡喃-8-基)乙酸
参照实施例35的合成方法制备本实施例化合物,不同的是使用2-(2-羟基-3-(3-(4-羟基-3-(3-甲基丁-2-烯基)苯基)丙烯酰基)苯基)乙酸120mg(0.33mmol)为起始原料,得到产品83mg,收率69.2%。
1H-NMR(DMSO-d6,400MHz):1.284(6H,s,-CH3×2),1.758-1.791(2H,t,J=6.8Hz,-CH2-CH),2.811-2.853(2H,dd,-CH2),3.118-3.193(1H,m,-CH2-CH),3.586(2H,s,-CH2-COOH),5.475-5.507(1H,dd,-OH),6.722-6.743(1H,d,J=8.4Hz,Ar-H),7.019-7.057(1H,m,Ar-H),7.189-7.232(2H,m,Ar-H×2),7.509-7.527(1H,dd,Ar-H),7.694-7.713(1H,dd,Ar-H),12.276(1H,s,-COOH).ESI-MS[M+1]:367.1。
实施例39:2-(2-(4-羟基-3-(3-甲基丁-2-烯基)苯基)-4-氧代苯并二氢吡喃-6-基)乙酸
参照实施例35的合成方法制备本实施例化合物,不同的是使用2-(4-羟基-3-(3-(4-羟基-3-(3-甲基丁-2-烯基)苯基)丙烯酰基)苯基)乙酸120mg(0.33mmol)为起始原料,得到产品81mg,收率67.8%。
1H-NMR(DMSO-d6,400MHz):1.283(6H,s,-CH3×2),1.756-1.988(2H,t,J=6.8Hz,-CH2-CH),2.739-2.755(3H,m,-CH2,-CH),3.227-3.316(1H,m,-CH2-CH),3.592(2H,s,-CH2-COOH),5.484-5.516(1H,dd,-OH),6.727-6.748(1H,d,J=8.4Hz,Ar-H),7.001-7.023(1H,d,J=8.8Hz,Ar-H),7.215-7.264(2H,m,Ar-H×2),7.447-7.474(1H,dd,Ar-H),7.669-7.674(1H,d,J=2Hz,Ar-H).ESI-MS[M+1]:367.2。
实施例40:2-(2-(4-羟基-3-(3-甲基丁-2-烯基)苯基-6-甲基-4-氧代苯并二氢吡喃-8-基)乙酸
参照实施例35的合成方法制备本实施例化合物,不同的是使用2-(2-羟基-3-(3-(4-羟基-3-(3-甲基丁-2-烯基)苯基)丙烯酰基)-5-甲基苯基)乙酸125mg(0.33mmol)为起始原料,得到产品84.2mg,收率67.2%。
1H-NMR(DMSO-d6,400MHz):1.173(6H,s,-CH3×2),1.753-1.786(2H,t,J=6.8Hz,-CH2-CH),2.273(3H,S,Ar-CH3),2.743-2.820(3H,m,-CH,-CH2),3.081-3.156(1H,m,-CH2-CH),5.419-5.452(1H,dd,Ar-OH),6.715-6.736(1H,d,J=8.4Hz,Ar-H),7.176-7.222(2H,m,Ar-H×2),7.341-7.346(1H,d,J=2Hz,Ar-H),7.497-7.500(1H,d,J=1.2Hz,Ar-H),12.311(1H,s,-COOH).ESI-MS[M+1]:381.2。
实施例36、37、44、45、46、47、48、49的合成参照实施例35的合成,如下面的表4。
表4:参照实施例35合成的部分化合物:
实施例41:2-(2-(4-(甲氧基甲氧基)-3-(3-甲基丁-2-烯基)苯基-4-氧代苯并二氢吡喃-8-基)乙酸
2-(2-羟基-3-(3-(4-(甲氧基甲氧基)-3-(3-甲基丁-2-烯基)苯基)丙烯酰基)苯基)乙酸123mg(0.3mmol),溶于6mL乙醇当中,加入乙酸钠180mg,滴入几滴水使乙酸钠完全溶解。回流反应2天。加入乙酸乙酯20mL稀释,用水15mL×2洗涤,有机相干燥,过滤浓缩,层析柱分离,二氯甲烷/甲醇(35:1),得到黄色固体33mg,收率26.8%。
1H-NMR(DMSO-d6,400MHz):1.684-1.690(6H,d,J=2.4Hz,-CH3×2),2.886-2.893(1H,dd,-CH2之一),3.105-3.137(1H,m,-CH2之一),3.283-3.383(7H,m,-CH2×3,-CH),3.599(2H,s,-CH2-COOH),5.244-5.272(3H,t,J=6.4Hz,-OCH3),5.537-5.569(1H,dd,-CH2-CH),7.044-7.069(2H,t,J=7.6Hz,Ar-H×2),7.276-7.296(2H,t,J=8.0Hz,Ar-H×2),7.518-7.537(1H,d,J=7.6Hz,Ar-H),7.692-7.711(1H,d,J=7.6Hz,Ar-H),12.329(1H,s,-COOH).ESI-MS[M+1]:411.1。
实施例42:2-(2-(4-(甲氧基甲氧基)-3-(3-甲基丁-2-烯基)苯基-4-氧代苯并二氢吡喃-6-基)乙酸
参照实施例41的合成方法制备本实施例化合物,不同的是使用2-(4-羟基-3-(3-(4-(甲氧基甲氧基)-3-(3-甲基丁-2-烯基)苯基)丙烯酰基)苯基)乙酸123mg(0.3mmol)为起始原料,得到产物31mg,收率25.2%。
1H-NMR(DMSO-d6,400MHz):1.683(6H,s,-CH3×2),2.746-2.795(1H,dd,-CH2-CH),3.286-3.380(6H,m,-CH2×3),3.591(2H,S,-CH2-COOH),5.237-5.256(3H,S,-CH3),5.542-5.567(1H,dd,-CH2-CH),7.008-7.029(2H,m,Ar-H×2),7.305-7.318(2H,m,Ar-H×2),7.451-7.478(1H,dd,Ar-H),7.664-7.669(1H,d,J=2Hz,Ar-H),12.291(1H,s,-COOH).ESI-MS[M+1]:411.1。
实施例43的合成参照实施例41的合成。
实施例53:2-(3-羟基-2-(4-羟基苯基)-4-氧代-4H-苯并吡喃-8-基)乙酸
2-(2-羟基-3-(3-(4-羟基苯基)丙烯酰基)苯基)乙酸0.25g(0.84mmol),溶于6mL甲醇当中。室温搅拌下,加入16%氢氧化钠水溶液(约为5N)1.2mL,体系颜色由黄变红。再加入30%双氧水0.15mL,体系颜色逐渐褪去,反应1小时,紫外365nM处显示强吸收。用冰水20mL稀释,然后酸化至pH 2,有大量明黄色固体析出,过滤,得到产物0.18g,收率69.2%。
1H-NMR(DMSO-d6,400MHz):3.989(2H,s,-CH2-COOH),6.924-6.947(2H,m,Ar-H×2),7.387-7.426(1H,t,Ar-H),7.696-7.717(1H,dd,Ar-H),8.012-8.094(3H,m,Ar-H×3),9.399(1H,s,-OH),10.126(1H,s,-OH),12.632(1H,s,-COOH)。ESI-MS[M+1]:313.1。
实施例54:2-(3-羟基-2-(4-羟基苯基)-4-氧代-4H-苯并吡喃-6-基)乙酸
参照实施例53的合成方法制备本实施例化合物,不同的是使用2-(4-羟基-3-(3-(4-羟基苯基)丙烯酰基)苯基)乙酸200mg(0.67mmol)为起始原料,得到产物0.16g,收率76.2%。
1H-NMR(DMSO-d6,400MHz):3.771(2H,s,-CH2-COOH),6.936-6.958(2H,d,J=2Hz,Ar-H×2),7.671-7.681(2H,t,J=2Hz,Ar-H×2),8.089-8.120(3H,d,J=8.4Hz,Ar-H×3),9.337(1H,s,Ar-OH),10.084(1H,s,-OH),12.433(1H,s,-COOH)。ESI-MS[M+1]:313.0。
实施例59:2-(3-羟基-2-(4-(甲氧基甲氧基)-3-(3-甲基丁-2-烯基)苯基-4-氧代-4H-苯并二氢吡喃-8-基)乙酸
参照实施例53的合成方法制备本实施例化合物,不同的是使用2-(2-羟基-3-(3-(4-(甲氧基甲氧基)-3-(3-甲基丁-2-烯基)苯基)丙烯酰基)苯基)乙酸200mg(0.5mmol)为起始原料,得到产物133.7mg,产率64.7%。
1H-NMR(DMSO-d6,400MHz):1.717-1.738(6H,d,J=8.4Hz,-CH3×2),3.350-3.414(5H,m,-CH2-CH;-OCH3),3.990(2H,s,-CH2-COOH),5.324-5.341(3H,m,-OCH2;-CH2-CH),7.193-7.215(1H,d,J=8.8Hz,Ar-H),7.397-7.435(1H,t,J=7.6Hz,Ar-H),7.708-7.730(1H,dd,Ar-H),8.019-8.061(3H,m,Ar-H×3),9.491(1H,s,-OH),12.617(1H,s,-COOH)。ESI-MS[M+1]:425.1。
实施例60:2-(3-羟基-2-(4-(甲氧基甲氧基)-3-(3-甲基丁-2-烯基)苯基)-4-氧代-4H-苯并吡喃-6-基)乙酸
参照实施例53的合成方法制备本实施例化合物,不同的是使用2-(4-羟基-3-(3-(4-(甲氧基甲氧基)-3-(3-甲基丁-2-烯基)苯基)丙烯酰基)苯基)乙酸200mg(0.5mmol)为起始原料,得到产物132.9mg,收率64.3%。
1H-NMR(DMSO-d6,400MHz):1.707-1.733(6H,d,J=10.4Hz,-CH3×2),3.314-3.410(5H,m,-CH2×2,-CH),3.773(2H,s,-CH2-COOH),5.275-5.326(3H,s,-OCH3),7.198-7.221(1H,d,Ar-H),7.686(2H,s,Ar-H×2),7.991-8.043(3H,m,Ar-H×3),9.404(1H,s,-OH),12.366(1H,s,-COOH).ESI-MS[M+1]:425.1。
实施例50、51、52、55、56、57、58、61、62、63、64、65、66、67的合成参照实施例53,如下面的表5。
表5:参照实施例53合成的部分化合物
药理学试验及活性数据
如下面的试验例1-3。
试验例1:AlphaLISA法检测RAW 264.7细胞培养上清内TNF-α浓度
实验方法如下:取对数生长期的RAW 264.7细胞,用含有10%胎牛血清的RPMI-1640培养基配制成细胞悬液,于96孔培养板内接种,每孔100μl(含2500个RAW264.7细胞),置于37℃,5%CO2温箱内,加入目标化合物作用一定时间后,将96孔板离心2000r、10min,之后每孔取150μl上清,分装到三个EP中,每支50μl,于-20℃冻存备用。随后按照AlphaLISA TNF-α试剂盒说明,配制标准曲线溶液,并取5μl上清接种于White OptiPlate-384孔板中,然后每孔加入AlphaLISA Anti-Analyte Acceptor beads(终浓度10μg/ml)和Biotinylated Antibody Anti-Analyte(终浓度1nM)混合液20μl(Acdeptor beads:Biotinylated Antibody Anti-Analyte:1X buffer=1:2:198),23℃孵育60min后加入Donor beads(终浓度为40μg/ml)25μl(Donor beads:1X buffer=1:61.5),23℃避光孵育30min后利用EnVision检测。
实验结果如表6。
试验例2:MTS法测定化合物的细胞毒作用
实验方法如下:取对数生长期的小鼠单核巨噬细胞株RAW 264.7细胞,用含有10%胎牛血清的RPMI-1640培养基配制成细胞悬液,接种于96孔培养板内,每孔100μl(含2500个肿瘤细胞),置于37℃,5%CO2温箱内。加入目标化合物后作用一定时间后,每孔加入MTS溶液10μl,37℃孵育4小时。用酶联免疫检测仪在490nm波长处测定其吸光值。
细胞存活率(%)=(给药组OD-空白组OD)/对照组OD×100%
实验结果如表6。
表6:部分化合物活性实验结果
“-”表示未进行检测。
目标化合物浓度为30μg/ml,LPS浓度为2.5ng/ml,以DMSO或DMSO与LPS为对照;“a”表示目标化合物单独作用促进RAW 264.7细胞分泌TNF-α的分泌率;“b”表示与LPS(脂多糖2.5ng/ml)合用时目标化合物与LPS合用促进RAW 264.7细胞分泌TNF-α的分泌率;“c”表示目标化合物单独作用对RAW 264.7细胞的生长抑制作用;“d”表示目标化合物与LPS合用对RAW 264.细胞的生长抑制作用。
结果显示,式I、式II化合物具有抗肿瘤活性,因此式I、式II化合物可作为抗肿瘤药物用于抗肿瘤的研究中,具有开发成为抗肿瘤药物的潜力。
此外,一般的促TNF-α分泌都是通过LPS途径,实验结果显示,本发明的化合物不依赖LPS途径也能够产生TNF-α促分泌作用。
试验例3:鸡胚绒毛尿囊膜模型考察化合物对血管的影响
实验方法如下:在无菌条件下,将10μl相应浓度目标化合物溶液均匀涂布于Whatman滤纸上,阴性对照涂布相同体积的0.3%DMSO溶液,将受精种蛋置于37℃、60%相对湿度的恒温孵育箱中进行孵化,6天后,将蛋胚下层的CAM膜暴漏。将载样滤纸置于CAM和卵黄囊膜处血管较多部位,然后用灭菌透明胶封口,继续培养至8天,除去覆盖在鸡胚上的透明胶布。对给药部位进行拍摄,处理图片,统计给药组和对照组血管面积,计算两组血管面积占CAM总面积的百分比(采用Adobe Photoshop CS5、Image-Pro Plus 6.0软件对鸡胚尿囊膜血管进行可视化和后续的面积定量,n=2)。
血管破坏率(Distrupting Ratio,%)=[1-(实验组血管面积/溶剂对照组血管面积)]×100%。
实验结果如表7。
表7:目标化合物对鸡胚绒毛尿囊膜血管的影响
目标化合物的浓度为10μg/鸡胚,以DMSO为对照。
实验结果显示,本发明的化合物可以促进肿瘤细胞分泌TNF-α,同时还能显著的减少鸡胚绒毛尿囊膜血管数目。因此,本发明的化合物能够用于制备抗肿瘤药物。
试验例4:对肺癌荷瘤小鼠肿瘤血管的破坏作用
实验步骤:
(1)小鼠LLC皮下荷瘤模型的建立
将实验室内Lewis肺癌细胞荷瘤的C57BL/6小鼠脱颈处死,75%酒精中浸泡消毒1~2min。之后在无菌操作台内分离其皮下瘤块,生理盐水冲洗2遍,放在事先经过高温灭菌的150~200目不锈钢筛网上,剪刀剪碎后充分研磨。筛网下置盛有适量无菌生理盐水的玻璃培养皿收集研碎后的细胞,并将收集到的所有细胞液合并于离心管中,1500rpm离心5min。弃上清,用适量无菌生理盐水重悬制成细胞悬液,充分吹打均匀,转移至25cm2培养瓶中,封口膜封口待用。
用75%酒精棉球擦拭消毒C57BL/6小鼠右侧腋下皮肤,1ml注射器吸取配好的细胞悬液注入小鼠腋下皮下,每只0.2ml(每只鼠在吸取细胞液时都注意充分混匀),停针2~3s后翻转针头拔针。接种完毕后,隔天记录各组小鼠的体重变化和瘤体生长情况。按公式V=D(长径)×d2(短径)/2计算瘤体体积。若动物发生死亡,记录其存活时间并视情况解剖观察其体内肿瘤侵染情况。
(2)给药与检测
待瘤体直径长至约10mm时,除去未成瘤动物。剩余所有成瘤鼠按瘤体大小进行适当调整,使每组样本尽量平均分配。试药组相应腹腔注射DMXAA(20mg/kg)、实施例2化合物(20mg/kg),对照组腹腔注射相应体积的1%DMSO。所有小鼠于给药3h后尾静脉注射荧光染料Hoechst 33342(800μg/mouse,0.1ml)。注射1min后将小鼠脱颈处死,剥离瘤体拍照、称重,计算瘤体体积,并迅速用锡箔纸包好,冻存于液氮中进行冷冻切片,于倒置荧光显微镜下观察。
实验结果:如附图1、2、3(3A、3B、3C)所示。
结果表明,实施例2化合物能破坏肿瘤血管,减少肿瘤血液灌注。
尽管本发明的具体实施方式已经得到详细的描述,本领域技术人员将会理解。根据已经公开的所有教导,可以对那些细节进行各种修改和替换,这些改变均在本发明的保护范围之内。本发明的全部范围由所附权利要求及其任何等同物给出。
- 还没有人留言评论。精彩留言会获得点赞!