配体‑温敏性离子液体共聚物及其制备方法和用途与流程
本发明属于催化聚合技术领域,涉及一种配体-温敏性离子液体共聚物,其制备方法,及其在ATRP反应中作为配体来回收及循环利用催化剂的用途。
背景技术:
离子液体(ionic liquid,IL)是由有机阳离子和有机/无机阴离子组成的,在常温下呈透明油状液态的有机盐,具有宽液程、低熔点、难挥发、易导电、热稳定等物理性质,通过改变阴/阳离子可以对上述性质进行调节,以满足不同的应用需求。由于种类繁多加之具有再生循环无污染的优异特性,离子液体在催化领域中的应用大放异彩。
聚离子液体(poly(ionic liquids),PILs)是由离子液体单体聚合而成的一类新兴材料,可以分为以下四类:1)聚阳离子型离子液体;2)聚阴离子型离子液体;3)聚两性型离子液体;4)共聚型离子液体。其中,针对聚阳离子型离子液体的研究较为广泛。在通常情况下,聚离子液体为固体状态或胶质,但其玻璃化转变温度(Tg)通常远低于常规的玻璃离子体,导致其在催化领域稍逊一筹。
通过对聚离子液体的结构和性能进行设计,使得其嵌段或无规共聚物集离子液体的优异特性与其他功能性单体的独特性质于一体。另外,也可通过活性聚合方法对PILs进行精密控制与合成,进而运用至其他领域。但相关研究最近十几年才开始蓬勃发展,因此如何对聚离子液体进行设计、扩大其种类并拓展其应用已经成为当今研究的热点之一。
原子转移自由基聚合方法(Atom Transfer Radical Polymerization,ATRP)是一种优异的活性聚合方法,具有单体适用面广、聚合方法多样、可对聚合物进行精密合成与设计等优点。但是,ATRP聚合方法中使用的金属盐催化剂会残留在聚合物中,使聚合物发生着色,这就限制了ATRP的应用,导致资源浪费,并且对环境造成破坏。
为了解决催化剂的残留问题,各种方法竞相发展。液/液两相分离法通过简单的分离过程来实现催化剂与产物的分离,在保证催化剂低残留的同时实现可控聚合,加之可对催化剂络合物进行回收及循环利用,使其从众多方法中脱颖而出。温控相分离催化(Thermo-regulated Phase Separable Catalysis,TPSC)通过改变温度来实现均相高效聚合、异相分离催化剂与聚合物以及对催化剂络合物的回收,但这种方法在经过几次回收后催化剂络合物的损失较严重或者影响体系的控制性和反应速率(参见M. Ding, Z. Cheng, Macromol. rapid comm., 2015, 36: 1702-1721)。
技术实现要素:
针对以上问题,本发明设计并合成了一种新型的配体-温敏性离子液体共聚物,其中离子液体单体为发展较为广泛的咪唑类离子液体,并且结构中包含聚乙二醇单甲醚链段,由于醚链上的氧原子与咪唑上的氢原子会产生分子内或分子间作用力,因此这种离子液体的粘度较低,并且表现出较为理想的温敏性;另外,共聚物中还包含一种具有配体功能的单体,其与离子液体单体通过自由基聚合,最终得到配体-温敏性离子液体共聚物PILL。上述方法将两种单体以共价键的形式连接在一起,根据这种无规共聚物的温敏性与配体功能,本发明人选择将其运用到ATRP聚合方法学中,在实现高效回收及循环利用催化剂的同时,以期解决配体的流失问题。
首先,本发明提供了一种配体-温敏性离子液体共聚物,其具有如式I所示的结构:
;
其中:x:y=1:5~7,优选1:6;MPEG表示聚乙二醇单甲醚片段。
其次,本发明提供了上述配体-温敏性离子液体共聚物的制备方法,其包括如下步骤:
(1)配体单体(MA-LN)的合成:
在氩气保护条件下,向溶解有三乙酰氧基硼氢化钠的四氢呋喃中依次加入乙醇胺、2-吡啶甲醛和冰醋酸,室温下反应3天,经纯化得到中间体II;其中:三乙酰氧基硼氢化钠、乙醇胺、2-吡啶甲醛、冰醋酸之间的摩尔比为4:1:2:3;
;
向四氢呋喃中加入中间体II和三乙胺,在冰盐浴及氩气保护条件下,滴加丙烯酰氯,室温下过夜反应,经纯化配体单体III;其中:中间体II、三乙胺、丙烯酰氯之间的摩尔比为1:3:2;
;
(2)离子液体单体(MA-IL)的合成:
向乙醚中加入聚乙二醇单甲醚和三乙胺,在冰水浴条件下,加入甲基磺酰氯,室温下反应2天,经纯化得到中间体IV;其中:聚乙二醇单甲醚、三乙胺、甲基磺酰氯之间的摩尔比为1:1:1;
;
在冰水浴条件下,向水中加入氢氧化钠和四乙基溴化铵,然后加入咪唑和中间体IV,室温下反应3天,经纯化得到中间体V;其中:氢氧化钠、四乙基溴化铵、咪唑、中间体IV之间的摩尔比为10:0.03:1.1:1;
;
在冰水浴及氩气保护条件下,向二氯甲烷中加入2-溴乙醇,然后依次滴加丙烯酰氯和三乙胺,室温下过夜反应,经纯化得到中间体VI;其中:2-溴乙醇、丙烯酰氯、三乙胺之间的摩尔比为1.1:1:1;
;
在氮气保护条件下,向乙腈中加入中间体V和中间体VI,45℃下反应4天, 经纯化得到离子液体单体VII;其中:中间体V、中间体VI之间的摩尔比为1:1;
;
(3)配体-温敏性离子液体共聚物(PILL)的合成:
将配体单体III、离子液体单体VII和偶氮二异丁腈加入到反应容器中,加入二甲基亚砜后除氧、密封,80℃下反应3天,经纯化得到配体-温敏性离子液体共聚物;其中:配体单体III、离子液体单体VII、偶氮二异丁腈之间的摩尔比为1:10:1;
。
优选的,在上述制备方法中,有机溶剂在使用之前经过无水处理。
最后,本发明提供了上述配体-温敏性离子液体共聚物在构建温控相分离体系并且在ATRP反应中作为配体来回收催化剂的用途。
优选的,在上述用途中,所述ATRP反应包括如下步骤:
(1)向反应容器中加入催化剂和配体-温敏性离子液体共聚物,再加入溶解有单体、引发剂、还原剂的有机溶剂,经除氧、密封,在温度为70~90℃,转速为1800~2000 rpm的条件下进行聚合反应3~20小时;其中:单体、引发剂、催化剂、配体-温敏性离子液体共聚物、还原剂之间的摩尔比为200:0.5~2:0.5~2:1~4:0.5~1.5,单体、有机溶剂之间的体积比为1:2~4;
(2)反应结束后,将反应容器冷却至室温,反应体系分成有机相和催化剂/配体-温敏性离子液体共聚物相,分离有机相并用四氢呋喃稀释,然后加入到环己烷中沉淀,经抽滤、真空干燥,得到所需的聚合物;
(3)向另一个反应容器中加入分液处理后剩余的催化剂/配体-温敏性离子液体共聚物相,再加入单体、引发剂、还原剂和有机溶剂,即可进行下一轮的聚合,实现催化剂和配体-温敏性离子液体共聚物的回收及循环利用;其中:单体、引发剂和还原剂的用量分别与其在步骤(1)中的用量相等。
优选的,步骤(1)中的催化剂为溴化铜。
优选的,步骤(1)中的单体选自甲基丙烯酸甲酯、苯乙烯、丙烯酸甲酯、丙烯酸叔丁酯中的任意一种,优选甲基丙烯酸甲酯。
优选的,步骤(1)中的引发剂为α-溴代苯乙酸乙酯。
优选的,步骤(1)中的还原剂为偶氮二异丁腈。
优选的,步骤(1)中的有机溶剂为苯。
优选的,步骤(1)中单体、引发剂、催化剂、配体-温敏性离子液体共聚物、还原剂之间的摩尔比为200:1:1:4:0.8;单体、有机溶剂之间的体积比为1:3。
优选的,步骤(1)中的聚合反应在温度为70℃,转速为2000 rpm的条件下进行6小时。
优选的,步骤(3)中的回收及循环利用的次数为10次。
由于上述技术方案的运用,本发明与现有技术相比具有下列优点:
(1)本发明的配体-离子液体共聚物不仅拓展了聚离子液体共聚物的种类,还具有多种优异特性,基于其温敏性和配体功能成功地构建了温控相分离体系,并运用于ATRP催化剂的回收及循环利用;
(2)液/液两相回收体系虽然能够有效地实现催化剂的回收及循环利用,但是这种方法不能适用于包含对水敏感或水溶性差的基质的体系中,本发明的温控相分离方法可以很好地解决这一问题;
(3)本发明的配体-离子液体共聚物构建的温控相分离体系通过改变温度便可实现高温均相高效聚合和低温异相分离,在引发高效聚合的同时,还可实现催化剂的回收及循环利用,并且在很大程度上降低了回收循环中的配体损失;
(4)本发明的配体-离子液体共聚物构建的温控相分离体系使得催化剂损失更少且催化剂回收效率更高,回收循环十次后,每次聚合物相催化剂残留量在1.5 ppm左右,催化剂回收效率仍然在95%以上。
附图说明
图1是实施例1中的化合物1的1H-NMR谱图。
图2是实施例1中的化合物2的1H-NMR谱图。
图3是实施例2中的化合物3的1H-NMR谱图。
图4是实施例2中的化合物4的1H-NMR谱图。
图5是实施例2中的化合物5的1H-NMR谱图。
图6是实施例2中的化合物6的1H-NMR谱图。
图7是实施例3中的化合物7的1H-NMR谱图。
图8是不同引发剂浓度下所得聚合物的流出曲线图。
图9是MMA聚合反应的动力学行为示意图。
图10是聚合物PMMA的核磁氢谱。
图11是聚合物PMMA的扩链结果示意图。
图12是聚合物PMMA的大分子质谱图。
图13是催化剂经过十次循环利用后的回收效率示意图。
具体实施方式
下面将结合附图及具体实施例来进一步描述本发明的技术方案。可以理解的是,这些附图及实施例仅用于解释本发明,而不旨在限制本发明的保护范围。另外,除非另有特殊说明,下列实施例中所使用的各种试剂、材料、仪器等均可通过常规的商业手段获得。
化学试剂:
1、配体单体合成:
2-吡啶甲醛(98%,百灵威科技有限公司);乙醇胺(>99%,北京安耐吉能源有限公司);三乙酰氧基硼氢化钠(97%,北京安耐吉能源有限公司);丙烯酰氯(97%,北京安耐吉能源有限公司);
2、离子液体单体合成:
MPEG-350(北京安耐吉能源有限公司);甲基磺酰氯(分析纯,中国医药(集团)上海化学试剂公司);咪唑(分析纯,中国医药(集团)上海化学试剂公司);四乙基溴化铵(>97%,中国医药(集团)上海化学试剂公司);氢氧化钠(分析纯,中国医药(集团)上海化学试剂公司);丙烯酰氯(97%,北京安耐吉能源有限公司);2-溴乙醇(>96%,上海阿达玛斯有限公司);
3、聚合反应:
单体:甲基丙烯酸甲酯(MMA)(>99%)、苯乙烯(St)(>99%)、丙烯酸甲酯(MA)(>99%)、丙烯酸叔丁酯(BA)(>99%)均来自中国医药(集团)上海化学试剂公司;
引发剂:α-溴代苯乙酸乙酯(EBPA)(98%,阿法埃莎化学有限公司)
催化剂:溴化铜(分析纯,中国医药(集团)上海化学试剂公司);
还原剂:偶氮二异丁腈(AIBN)(分析纯,西格玛奥德里奇公司);
其他试剂:甲醇(工业级)、四氢呋喃(分析纯)、冰醋酸(分析纯)、三乙胺(分析纯)、二甲亚砜(分析纯)、二氯甲烷(分析纯)、无水乙醚(分析纯)、苯(分析纯)均来自中国医药(集团)上海化学试剂公司。
测试仪器及条件:
凝胶渗透色谱(GPC):日本东曹公司(TOSOH)HLC-8320型GPC;测试条件:Tskgel Super MultiporeHZ-N(4.6*150)两柱联用,示差检测器,流动相为四氢呋喃,流速为0.35 mL/min,柱温为40℃;
核磁共振(NMR):Bruker 300MHz核磁仪,溶剂为CDCl3或DMSO-d6;
基质辅助激光解吸电离飞行时间质谱(MALDL-TOF):配备1 kHz智能波束-II激光束的Bruker ultrafleXtreme型MALDL-TOF质谱仪。
实施例1:配体单体的合成。
在氩气保护条件下,向溶解有三乙酰氧基硼氢化钠(16.9 g,0.08 mol)的THF(300 mL)中依次缓慢加入分别溶解在THF(20 mL)中的乙醇胺(1.22 g,0.02 mol)、2-吡啶甲醛(4.3 g,0.04 mol)和冰醋酸(3.4 mL,0.06 mol),室温下反应三天,过滤,加二氯甲烷(30 mL)溶解后,用碳酸钠饱和溶液(3*30 mL)萃取,旋蒸除去THF后,采用柱层析提纯,得到淡黄色液态中间体1,其1H-NMR谱图如图1所示;
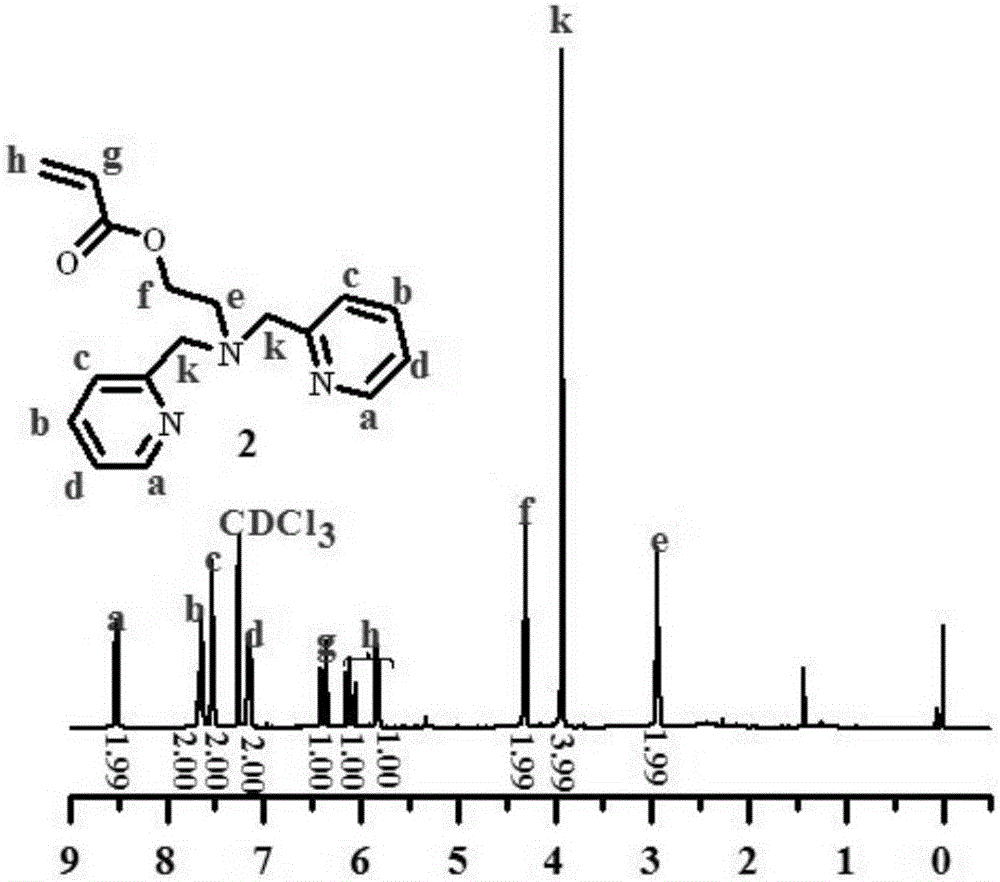
向THF(100 mL)中加入中间体1(4.8 g,2 mmol)和三乙胺(8.3 mL,6 mmol),在冰盐浴及氩气保护条件下,滴加溶解在THF(10 mL)中的丙烯酰氯(3.2 mL,4 mmol),室温下过夜反应,过滤,旋蒸除去THF后,采用柱层析提纯,得到淡黄色液态配体单体2,其1H-NMR谱图如图2所示。
实施例2:离子液体单体的合成。
向无水乙醚(300 mL)中加入MPEG-350(70 g,0.20 mol)和三乙胺(31.0 mL,0.20 mol),在冰水浴条件下,加入甲基磺酰氯(25.1 g,0.20 mol),室温下反应两天,过滤,旋蒸除去无水乙醚,加入二氯甲烷(100 mL)溶解后,用水(3*50 mL)萃取后分离有机相,旋蒸除去二氯甲烷,得到无色液态中间体3,其1H-NMR谱图如图3所示;
在冰水浴条件下,向水(300 mL)中加入氢氧化钠(56 g,1.4 mol)和四乙基溴化铵(0.88 g,4.2 mmol),大部分碱溶解后,加入咪唑(7.6 g,0.16 mol)和中间体3(50 g,0.14 mol),室温下反应三天,用二氯甲烷(4*100 mL)萃取后分离有机相,旋蒸除去二氯甲烷,得到无色液态中间体4,其1H-NMR谱图如图4所示;
在冰水浴及氩气保护条件下,向(100 mL)无水二氯甲烷中加入2-溴乙醇(28 g,0.22 mol),在30 min内滴加溶解在二氯甲烷(20 mL)中的丙烯酰氯(18.1 g,0.20 mol),持续搅拌30分钟,在60分钟内缓慢滴加溶解在二氯甲烷(20 mL)中的三乙胺(22 g,0.20 mol),室温下过夜反应,过滤,用去离子水(5*50 mL)萃取后分离有机相,旋蒸除去二氯甲烷,得到淡黄色液态中间体5,其1H-NMR谱图如图5所示;
在氮气保护条件下,向无水乙腈中加入中间体4(57.9 g,0.1 mol)与中间体5(17.9 g,0.1 mol),45℃下反应四天,旋蒸除去乙腈,粗产物加少量甲醇溶解,用无水乙醚(5*50 mL)萃取后分离有机相,旋蒸除去乙醚,得到橙色离子液体单体6,其1H-NMR谱图如图6所示。
实施例3:PILL的合成。
将配体单体2(0.297 g,1 mmol)、离子液体单体6(5.78 g,10 mmol)和引发剂AIBN(0.164 g,1 mmol)加入到25 mL Schlenk管中,加入DMSO(10 mL)后除氧、密封,转移至80℃油浴锅中反应三天,此时溶液由透明橙色变为棕色;反应结束后,反应物用少许甲醇溶解,用无水乙醚(100 mL)沉淀五次,真空烘干,得到粘稠状胶质共聚物7(PILL),其1H-NMR谱图如图7所示,经积分计算可知,配体片段与温敏性离子液体片段之间的摩尔比约为1:6。另外,对所得共聚物进行GPC测试,由其可知共聚物PILL的分子量为26300 g/mol。
实施例4:MMA的TPSC & ICAR ATRP反应及影响因素考察。
MMA的TPSC & ICAR ATRP反应过程如下所述:
将催化剂CuBr2(10.52 mg,0.0472 mmol)和实施例3中得到的作为配体的配体-温敏性离子液体共聚物(0.348 mg,0.189 mmol)加入到5 mL安瓿瓶中,再加入溶解有单体MMA(1.0 mL,9.44 mmol)、引发剂EBPA(8.25 μL,0.0472 mmol)和还原剂AIBN(6.22 mg,0.0379 mmol)的苯(3 mL),经除氧、密封,将安瓿瓶转移到油浴锅中,在70℃和2000 rpm的条件下进行聚合反应6小时;反应结束后,将安瓿瓶取出并置于冰水中,以便使反应体系冷却分层,分离有机相并用THF(4 mL)稀释,然后加入到正己烷(200 mL)中沉淀,经抽滤、真空干燥,得到所需的聚合物PMMA。
TPSC & ICAR ATRP反应的影响因素考察如下所述:
(1)不同用量的共聚物PILL对TPSC & ICAR ATRP反应的影响:
由于共聚物PILL在反应中不仅扮演配体的角色,而且还是TPSC两相中的温控相,因此PILL的用量对反应速率和控制效果都起到至关重要的作用。随着PILL用量的增加,在聚合温度下可以更快地形成均相,使得反应速率增加;与此同时,配体片段用量的相应增加也会在一定程度上提高催化剂在有机相中的溶解度,从而提高反应速率和反应的可控性。不同用量的PILL的影响如表1所示。
由表1可知,随着PILL用量的增加,反应速率相应增加,并且理论分子量与GPC 分子量更为接近,PDI也更窄,表明控制性更好。因此,选择用量为[MMA]0:[PILL]0=200:4的共聚物PILL作为配体。
(2)不同用量的溶剂苯对TPSC & ICAR ATRP反应的影响:
在反应过程中,反应速率会随着溶剂用量的增加而降低,其原因在于共聚物PILL为胶质,使得体系粘度较大,导致聚合物链的移动相对困难。因此,不同用量的溶剂也会影响聚合物的控制性能。不同用量的苯的影响如表2所示。
由表2可知,当采用不同体积的苯作为TPSC的另一相时,随着苯的体积的增加,反应速率有所减慢,但对聚合的控制效果来说,影响不大,都在可控范围以内。由于体系粘度较大,为减轻聚合后期高分子链的移动问题,最终选定3 mL的苯作为TPSC & ICAR ATRP的另一相。
(3)不同用量的催化剂与还原剂对TPSC & ICAR ATRP反应的影响:
通常,反应速率会随着催化剂用量的增加而增加,聚合控制效果也会变好。还原剂用量在一定程度上的增加会更快地将催化剂还原成低价,起初有大量引发种引发聚合,所以会导致聚合速率增加而控制性变差。不同用量的催化剂与还原剂的影响如表3所示。
由表3可知,随着催化剂用量的增加,反应速率呈现出“先增加后减少”的趋势,而聚合控制效果变得越来越好;而随着还原剂用量的增加,反应速率越来越快,控制性却越来越差。在保证聚合可控的同时考虑其反应速率,最终选定催化剂与还原剂的用量比例为[MMA]0:[CuBr2]0:[AIBN]0=200:1:0.8。
(4)不同用量的引发剂对TPSC & ICAR ATRP反应的影响。
通过改变初始的单体与引发剂的摩尔比,可以改变聚合物的分子量,从而实现聚合物的精密可控合成。在不同引发剂浓度下平行制备一系列聚合物,考察不同用量的引发剂对TPSC & ICAR ATRP反应的影响,其结果如表4所示。
由表4可知,在不同引发剂浓度下引发的TPSC & ICAR ATRP体系均可以得到可控的聚合,随着引发剂浓度的降低,反应速率变慢,PDI变窄,但是理论分子量与实际分子量之间的差距变大。
如图8所示,各种引发剂浓度下所得聚合物的流出曲线图皆成单峰正态分布。为了平衡聚合反应速率与聚合物的末端功能化度,最终选择[MMA]0:[EBPA]0=200:1作为动力学研究。
实施例5:聚合反应的动力学行为研究。
将不同时间得到的聚合物PMMA真空干燥,用重量法算出其转化率,并在相应的时间间隔进行三次催化回收循环利用的动力学研究,在日本东曹公司(TOSOH)HLC-8320型GPC上测试其分子量与分子量分布指数。
如图9(a)所示,每一次回收聚合物的转化率与时间都成线性关系,这表明体系中的自由基浓度保持恒定。如图9(b)所示,随着转化率的增加,理论分子量与实际分子量有一点偏差,这是由于聚合后期体系粘度较大,高分子链移动较为困难所致。但是,每一次回收所得聚合物的分子量都随着转化率的增加而增加,PDI也在可控范围以内随着分子量的增加而减小。由此可知,发明的催化体系具有活性可控的特征。
实施例6:聚合物PMMA的结构表征。
对如图10所示的PMMA的1H-NMR谱图(以 DMSO-d6作为溶剂,以TMS作为内标)进行分析可知,化学位移(δ)为4.10 ppm以及7.10~7.40 ppm处的信号峰对应引发剂EBPA中次甲基的氢原子与苯环的氢原子;由于溴原子具有吸电子诱导效应,使得聚合物链末端甲氧基的氢原子的化学位移出现偏移,对应3.78 ppm处的信号峰;其他氢原子也都有相应的信号峰,这说明EBPA引发剂片段成功地接入聚合物中,进一步证明了体系具有活性可控特征。
实施例7:聚合物PMMA的扩链表征。
由于PMMA的末端具有活性,因此可以作为大分子引发剂来引发新的聚合反应。如图11所示,以PMMA(GPC分子量为10600 g/mol,PDI为1.28)作为大分子引发剂来引发5 h的反应后,得到扩链PMMA(GPC分子量为23600 g/mol,PDI为1.31)。成功的扩链反应再次证明了PMMA的活性。
实施例8:聚合物PMMA的大分子测试表征。
如图12所示,PMMA(GPC分子量为6700 g/mol,PDI为1.22)通过MALDL-TOF质谱仪所测得的结构表明,通过与钠元素的结合,该聚合物存在唯一以溴封端的大分子结构。
实施例9:催化剂的循环利用、催化剂的残留以及催化剂的回收效率。
为了降低催化剂与配体的损失,本发明通过共价键将配体与温敏性离子液体结合成共聚物PILL,并运用于催化剂的回收及循环利用。聚合反应结束后,待有机层与催化层分离,移取一定量的上层有机相,高温灼烧,硝酸溶解过夜后,在容量瓶中用去离子水稀释成一定浓度的水溶液,经电感应耦合等离子体(ICP)测试后,计算可得苯相中的铜离子含量。苯相中的铜离子与初始PILL相的铜离子含量的百分比即为回收效率。
由表5可知,经过十次回收,PILL与催化剂仍具有大致不变的反应速率与控制效果。通过ICP测试可知,每一次回收中有机相残留的铜离子含量在1.5 ppm左右,很好地降低了聚合物中的催化剂残留。该结果表明,经过10次使用后的PILL与催化剂并没有降低活性,聚合过程一直可控,并且反应速率、控制效果以及催化剂在聚合物中的残留量都相差无几,证明该体系具有较高的反应活性与催化剂回收效率。
此外,如图13所示,即使经过十次回收及循环利用,催化剂的回收效率仍旧大于95%(相对于初始的铜离子含量),这是目前在ATRP催化剂的回收体系中回收效率最高的一个体系,同时也证明了本发明的配体-离子液体共聚物能够有效降低ATRP反应中催化剂和配体的损失。
实施例10:单体适用性的探究
本发明选择油溶性单体苯乙烯、丙烯酸甲酯和丙烯酸叔丁酯进行单体的适用性探索,不同单体需设定不同的聚合温度和时间,其结果如表6所示。
由表6可知,在设定时间和温度下进行不同单体的反应,所得聚合物的理论分子量与实际分子量都比较接近,并且分子量分布PDI也在可控范围之内。这表明该体系同样适用于苯乙烯、丙烯酸甲酯和丙烯酸丁酯等油溶性单体。
- 还没有人留言评论。精彩留言会获得点赞!