一种2‑位噻吩基取代的非对称型氟硼络合二吡咯甲川衍生物及其制备方法与流程
本发明涉及一种2-位噻吩基取代的非对称型氟硼络合二吡咯甲川衍生物及其制备方法,该类衍生物可应用于荧光染料、发光材料、光伏材料、生命科学、分析科学、环境能源科学、有机太阳能电池等领域。
背景技术:
氟硼络合二吡咯甲川(BODIPY)衍生物是一类良好的光敏染料,具有良好的稳定性,高的摩尔消光系数 (1×105 M-1 cm-1) 和较高的氧化电位,并且其可以修饰的位置多,可通过选择性的接入具有相应功能的基团来调节BODIPY染料的吸收/发射波长等光学性质、稳定性、以及溶解性等物理性质。自1968年被首次报道以后就受到人们的关注,近二十年来更是被广泛研究,已被运用到荧光染料、发光材料、光伏材料、生命科学、分析科学、环境科学、能源科学、有机太阳能电池等领域。
在小分子太阳能电池给体材料中,分子一般都是对称的结构,非对称的D-A分子还很少有文献报道。为了改良一些对称的D-A分子的光谱吸收性质,本发明设计了一种以BODIPY为中心吸电子基元,2,6-位分别引入噻吩和芴、咔唑以及三苯胺等基团,构建了一系列结构新颖的非对称BODIPY衍生物,并研究它们的光物理性质、电化学行为,以揭示这类分子结构与性质之间的关系。
研究结果表明,这类分子的吸收光谱宽阔,摩尔消光系数高,分子内电荷迁移和能量转移均较为顺畅,适合构建一些有机光电子材料,如有机太阳能电池材料等。
技术实现要素:
本发明的目的是提供一种2-位噻吩基取代的非对称D-A型氟硼络合二吡咯甲川(BODIPY)衍生物,其具有较为稳定、宽阔、强烈的光谱吸收,材料在空气中稳定性好。
本发明的另一个目的是提供一种2-位噻吩基取代的非对称型氟硼络合二吡咯甲川衍生物的制备方法,其制备成本低、反应条件温和、易于控制。
为实现上述目的,本发明采用以下技术方案:一种2-位噻吩基取代的非对称型氟硼络合二吡咯甲川衍生物,具有通式Ⅰ的化学结构:
Ⅰ
式中,
n为1-20的自然数。
一种2-位噻吩基取代的非对称型氟硼络合二吡咯甲川衍生物的制备方法,包括以下步骤:
(1) 2-碘-9,9-二甲基芴与双联频哪醇硼酯反应,得到中间体1,其结构为:
(2) 3-溴-9-辛基咔唑与双联频哪醇硼酯反应,得到中间体2,其结构为:
(3) 4-溴-N,N-二{4-[2-(2-甲基庚基)]苯基}苯胺与双联频哪醇硼酯反应,得到中间体3,其结构为:
(4)对羟基苯甲醛与溴代烷烃反应,然后再与新蒸吡咯和三氟化硼-乙醚反应,最后再经N-溴代丁二酰亚胺溴化,得到中间体4,其结构为:
(5) 中间体4和2-(三丁基锡)噻吩通过Stille偶联得到中间体5,其结构为:
(6) 中间体1和中间体5在催化剂的催化下反应生成目标产物BDP1,其结构为:
(7) 中间体2和中间体5 在催化剂的催化下反应生成目标产物BDP2,其结构为:
(8) 中间体3和中间体5在催化剂的催化下反应生成目标产物BDP3,其结构为:
作为上述技术方案的优选,步骤(1)-(8)中所述反应的反应介质为正己烷、四氢呋喃、N,N-二甲基甲酰胺、二氯甲烷、甲苯、氯仿、石油醚、乙酸乙酯、乙醇的一种或几种混合。
作为上述技术方案的优选,步骤(4)、(5)、(6)、(7)、(8)中所述催化剂为四(三苯基膦)钯、双(三苯基膦)二氯化钯、三(二亚苄基丙酮)二钯、[1,1'-双(二苯基磷)二茂铁]二氯化钯、 三氯化铟中的一种或几种混合。
作为上述技术方案的优选,步骤(5)中,合成中间体5的反应中,中间体4和2-(三丁基锡)噻吩的摩尔比为1:1.1-1:2。
作为上述技术方案的优选,步骤(5)中,所述反应的反应温度为90-150℃。
作为上述技术方案的优选,步骤(6)、(7)、(8)中,所述反应的反应温度为60-120℃。
作为上述技术方案的优选,步骤(5)-(8)中,所述反应的反应时间为12-36h。
与现有技术相比,本发明具有以下有益效果:
(1)本发明通过一系列反应合成中间体5,最后再利用该中间体与多种修饰基团进行Suzuki/Stille偶联反应,得到2-位噻吩基取代,6-位分别为芴、咔唑和三苯胺等强给电子单元取代的目标染料分子。
(2)本发明提供的合成方法制得的目标产物合成成本低、反应温和、易于控制。
(3)通过对几种目标产物的光谱和电化学数据分析,可以看出这些分子的吸收光谱宽阔,摩尔消光系数高。电化学测试结果显示:几种目标产物的HOMO能级分别在-5.2 ~ -5.4 eV之间,LUMO能级在-3.3 ~ -3.4 eV。
附图说明:
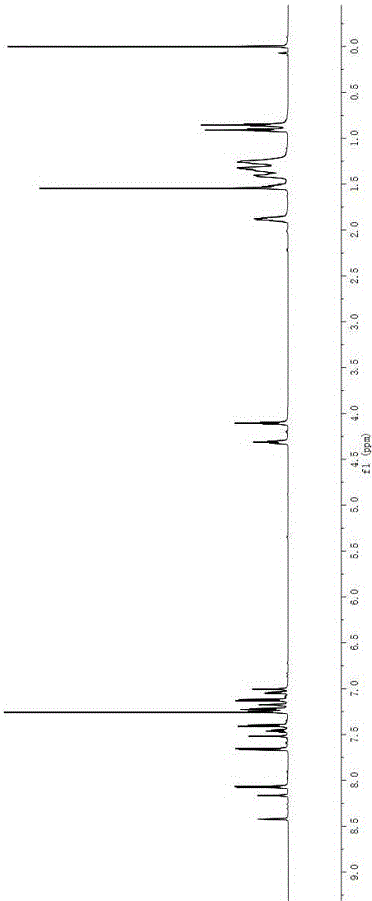
图1为本发明实施例1的BDP11HNMR图。
图2为本发明实施例1的BDP21HNMR图。
图3为本发明实施例1的BDP31HNMR图。
图4为本发明实施例1的BDP113C NMR图。
图5为本发明实施例1的BDP213C NMR图。
图6为本发明实施例1的 BDP1质谱图。
图7为本发明实施例1的BDP2质谱图。
图8为本发明实施例1的BDP3质谱图。
具体实施方式:
为更好的理解本发明,下面通过实施例及附图对本发明进一步说明,实施例只用于解释本发明,不会对本发明构成任何的限定。
实施例1
中间体1的合成:
在2-碘-9,9’-二甲基芴 (3.2 g, 10 mmol)、双联频哪醇硼酯 (2.79 g, 11 mmol)、乙酸钾 (2.94 g, 30 mmol)、DMF (80 mL)的混合液中加入Pd(dppf)Cl2 (360 mg, 0.5 mmol)。体系在氩气保护下, 90oC反应8 h。停止反应,混合物冷却至室温后倒入200 mL 水中,用乙酸乙酯萃取 (50 mL × 3),再用饱和食盐水洗涤数次,合并有机相,无水硫酸镁干燥。减压旋转蒸发,去除溶剂,残余物以石油醚:乙酸乙酯 = 20:1 (v/v) 为洗脱剂用硅胶 (200-300目) 进行柱层析分离,得到白色固体2.82 g,产率88%。1H NMR (600 MHz, CDCl3) δ: 7.91 (s, 1H), 7.85 (d, J = 7.5 Hz, 1H), 7.76 (t, J = 6.8 Hz, 2H), 7.48-7.45 (m, 1H), 7.37-7.34 (m, 2H), 1.53 (s, 6H), 1.40 (s, 12H)。
中间体2的合成:
在3-溴-9-辛基咔唑 (2 g, 5.6 mmol)、双联频哪醇硼酯 (1.7 g, 6.7 mmol)、乙酸钾 (2.7 g, 28 mmol)、DMF (80 mL)的混合液中加入Pd(dppf)Cl2 (150 mg, 0.21 mmol)。体系在氩气保护下, 90oC反应8 h。停止反应,混合物冷却至室温后倒入200 mL 水中,用二氯甲烷萃取 (50 mL × 3),再用饱和食盐水洗涤数次,合并有机相,无水硫酸镁干燥。减压旋转蒸发,去除溶剂,残余物以石油醚:乙酸乙酯 = 20:1 (v/v) 为洗脱剂用硅胶 (200-300目) 进行柱层析分离,得到白色固体1.7 g,产率76%。1H NMR (600 MHz, CDCl3), δ: 8.60 (s, 1H), 8.13 (d, J = 7.6 Hz, 1H), 7.92 (d, J = 8.1 Hz, 1H), 7.45 (d, J = 7.3 Hz, 1H), 7.40 (t, J = 9.5 Hz, 2H), 7.23 (d, J = 7.0 Hz, 1H), 4.30 (t, J = 6.9 Hz, 2H), 1.90-1.86 (m, 2H), 1.44 (s, 12H), 1.36-1.28 (m, 10H), 0.85 (t, J = 13.6 Hz, 3H). MALDI-TOF-MS, m/z: calcd for C26H36BNO2 [M]+: 405.300; found 405.333。
中间体3的合成:
中间体3 的反应条件与合成中间体2的条件类似,用4-溴-N,N-二(4-异辛基苯基)苯胺作为反应底物。纯化后得到白色固体, 产率84%。1H NMR (600 MHz, CDCl3), δ: 7.64 (d, J = 8.5 Hz, 2H), 7.25 (d, J = 8.6 Hz, 4H), 7.01 (d, J = 8.6 Hz, 4H), 6.98 (d, J = 8.5 Hz, 2H), 1.71 (s, 4H), 1.42-1.35 (m, 12H), 1.33 (s, 12H), 0.78-0.71 (m, 18H)。
中间体4的合成:
在100 mL单口瓶中加入对辛氧基苯甲醛 (2.34 g, 10 mmol) 和新蒸的吡咯 (30 mL, 430 mmol), 向溶液中鼓氩气置换10 min,在氩气保护下迅速加入催化剂InCl3 (0.11 g, 0.5 mmol), 在室温下磁力搅拌5 h, 向其中加入NaOH (0.2 g, 5 mmol)粉末继续搅拌30 min,终止反应。减压蒸馏,回收多余的吡咯,得粗产品,用硅胶(200-300目)柱层析[洗脱液,V(石油醚):V(乙酸乙酯) = 7:1],纯化得到白色晶体2.45 g, 产率70%。
在500 mL三口瓶中,取上述合成的白色晶体 (3.50 g, 10 mmol) 溶解于二氯甲烷 (80 mL) 中,加入四氯苯醌 (2.9 g, 12 mmol), 室温条件下磁力搅拌,充分氧化8 h。然后将反应混合液置于氩气保护下,慢慢滴加三氟化硼乙醚 (37 mL, 300 mmol),搅拌反应10 min 后再缓慢加入三乙胺 (41.7 mL, 300 mmol),滴加完毕后继续反应8 h。混合物倒入氢氧化钠溶液中,用二氯甲烷萃取。有机相用无水硫酸钠干燥。过滤、滤液减压下去除溶剂。粗产品用硅胶 (200-300目) 柱层析[洗脱液,V(石油醚):V(乙酸乙酯)=10:1] 分离得红绿色粉末状化合物2.50 g,产率63%。
向100 mL三口瓶中依次加入上一步合成的红绿色固体 (1.0 g, 2.5 mmol)、DMF (20 mL) 和二氯甲烷 (20 mL),在室温下磁力搅拌几分钟后,装上恒压滴液漏斗,抽真空,通入氩气保护,将NBS (0.89 g, 5.0 mmol) 溶解在DMF (5 mL) 和二氯甲烷 (5 mL)中,通过恒压滴液漏斗逐滴滴入到三口瓶中,滴加完毕后再继续反应4 h,整个过程避光。向三口瓶中加入50 mL去离子水,用二氯甲烷萃取三次 (80 mL × 3),有机层再经饱和食盐水洗涤 (100 mL × 3),合并有机相,无水硫酸镁干燥过夜。过滤收集滤液,减压下旋转蒸发去除溶剂,得粗产品,用硅胶(200-300目)柱层析[洗脱液,V(石油醚):V(乙酸乙酯) = 20:1]纯化得红色粉末状固体 0.97 g,产率70%。1H NMR (600 MHz, CDCl3), δ: 7.84 (d, J = 6.4 Hz, 2H), 7.53 (d, J = 8.6 Hz, 2H), 7.07 (d, J = 8.7 Hz, 2H), 7.02 (s, 2H), 4.09 (t, J = 6.5 Hz, 2H), 1.92–1.82 (m, 2H), 1.52 (m, 10H), 0.91 (t, 3H). 13C NMR (151 MHz, CDCl3), δ: 162.72, 147.50, 143.22, 134.70, 132.93, 131.16, 125.48, 114.46, 106.99, 68.25, 30.91, 29.70, 29.31, 28.78, 25.61, 22.42, 13.93. MALDI-TOF-MS, m/z: calcd for C23H25BBr2F2N2O [M-47]+: 554.000; found 507.100。
中间体5的合成:
在100 mL单口瓶中,依次加入中间体4 (0.55 g, 1.0 mmol)、2-(三丁基锡)噻吩(0.45 g, 1.2 mmol), 氟化铯 (0.3 g, 2.0 mmol) 和甲苯 (30 mL),抽真空,再向反应瓶中加入四(三苯基膦)钯 (0.15 g, 0.13 mmol),氩气保护,110oC下磁力搅拌反应24 h。停止反应,冷却至室温,倒入水中,用乙酸乙酯萃取,再用饱和食盐水洗涤 (30 mL × 3),收集有机相,无水硫酸钠干燥。减压下旋转蒸发去除溶剂,得粗产品,用硅胶 (300-400目) 柱层析 [洗脱液,V(石油醚):V(乙酸乙酯) = 10:1],得到褐色粉末状化合物固体0.31 g,产率56%。1H NMR (400 MHz, CDCl3), δ: 8.25 (s, 1H), 7.82 (s, 1H), 7.60 (d, J = 3.6 Hz, 2H), 7.23 (s, 1H), 7.11-7.09 (m, 5H), 6.98 (s, 1H), 4.12 (t, J = 2.4 Hz, J = 2.8 Hz, 2H), 1.89 (m, 2H), 1.63-1.35 (m, 10H), 0.95 (t, 3H). MALDI-TOF-MS, m/z: calcd for C27H28BBrF2N2OS [M]+: 556.117; found 556.073。
BDP1的合成
在100 mL 单口瓶中,依次加入中间体1 (96 mg, 0.3 mmol) 、 中间体5 (140 mg, 0.25 mmol)、四(三苯基膦)钯 (11 mg, 0.01 mmol)、甲苯 (30 mL) 和碳酸钾溶液 (20 mL 2M),排除空气,氩气保护,混合物在分别在70℃、90℃、110℃下反应12h、24h、36h后停止反应。将反应混合液冷却至室温,倒入100 mL 蒸馏水中,用乙酸乙酯萃取数次,有机层再用饱和食盐水洗涤 (50 mL×3),合并有机相,无水硫酸镁干燥。减压下旋转蒸发去除溶剂。残余物以石油醚:乙酸乙酯 = 10: 1 (v/v)为洗脱剂用硅胶 (300-400目) 进行柱层析分离提纯,得到墨绿色固体204 mg,产率61%。1H NMR (600 MHz, CDCl3), δ: 8.35 (s, 1H), 8.16 (s, 1H), 7.72 (d, J = 7.6 Hz, 2H), 7.64 (d, J = 8.6 Hz, 2H), 7.58 (s, 1H), 7.52 (d, J = 9.2 Hz, 1H), 7.44 (d, J = 6.7 Hz, 1H), 7.36–7.31 (m, 2H), 7.22 (d, J = 5.0 Hz, 1H), 7.20–7.16 (m, 2H), 7.12 (d, J = 8.6 Hz, 2H), 7.06–7.02 (m, 1H), 7.00 (s, 1H), 4.10 (t, J = 6.5 Hz, 2H), 1.90–1.85 (m, 2H), 1.51 (s, 7H), 1.43–1.26 (m, 11H), 0.91 (t, J = 6.9 Hz, 3H). 13C NMR (151 MHz, CDCl3), δ: 161.92, 154.47, 153.78, 146.41, 142.03, 140.53, 132.48, 127.91, 127.39, 127.08, 125.23, 124.17, 123.20, 122.63, 120.51, 120.03, 119.62, 114.78, 68.44, 46.93, 31.83, 29.46, 27.17, 26.08, 22.68, 14.13. MALDI-TOF-MS, m/z: calcd for C42H41BF2N2OS [M]+:670.300; found 670.209. HRMS, m/z: calcd for C42H41BF2N2OS [M]+: 670.3001; found 670.2997。
BDP2的合成
向100ml圆底烧瓶中加入中间体2和中间体5,反应条件和合成BDP1的条件相同,纯化得到墨绿色固体,产率63%。1H NMR (600 MHz, CDCl3), δ: 8.42 (s, 1H), 8.17 (s, 1H), 8.07 (d, J = 7.9 Hz, 2H), 7.66 (d, J = 8.6 Hz, 2H), 7.52 (s, 1H), 7.46 (t, J= 7.6 Hz, 1H), 7.40 (d, J = 8.1 Hz, 2H), 7.25–7.21 (m, 3H), 7.18 (d, J = 4.3 Hz, 1H), 7.12 (d, J = 8.6 Hz, 2H), 7.06–7.03 (m, 1H), 7.01 (s, 1H), 4.31 (t, J = 7.3 Hz, 2H), 4.11 (t, J = 6.5 Hz, 2H), 1.88 (td, J = 12.7, 6.8 Hz, 4H), 1.44–1.23 (m, 20H), 0.91 (t, J = 6.9 Hz, 3H), 0.86 (t, J = 7.0 Hz, 3H). 13C NMR (151 MHz, CDCl3), δ: 161.92, 146.35, 142.39, 141.05, 140.88, 140.34, 136.13, 132.51, 130.15, 127.92, 125.81, 124.14, 123.18, 120.86, 120.33, 116.87, 114.78, 108.80, 31.83, 29.39, 29.04, 27.35, 26.10, 22.65, 14.11. MALDI-TOF-MS, m/z: calcd for C47H52BF2N3OS [M]+: 755.389; found 755.345. HRMS, m/z: calcd for C47H52BF2N3OS [M]+: 755.3892; found 755.3883。
BDP3的合成
向100ml圆底烧瓶中加入中间体3和中间体5,反应条件和合成BDP1的条件相同,纯化得到墨绿色固体,产率59%。1H NMR (400 MHz, CDCl3), δ: 8.03 (s, 2H), 7.56 (d, J = 8.5 Hz, 2H), 7.27 (d, J = 8.1 Hz, 13H), 7.02 (dd, J = 18.6, 8.4 Hz, 12H), 6.93 (d, J = 8.6 Hz, 4H), 4.06 (t, J = 6.5 Hz, 2H), 1.71 (s, 8H), 1.55 (s, 11H), 1.28 (d, J = 19.8 Hz, 18H), 0.75 (s, 42H). MALDI- TOF-MS, m/z: calcd for C61H74BF2N3OS [M]+: 945.561; found 945.258. HRMS, m/z: calcd for C61H74BF2N3OS [M]+: 945.5614; found 945.5611。
上述实施例中目标产物BDP1-3在CH2Cl2溶液中和固体膜上的紫外可见吸收光谱和荧光光谱结果见表1,实施例中目标产物BDP1-3电化学性质的相关数据见表2。
。
a measured in CH2Cl2 solution. b measured in the neat film。
表1结果显示,在CH2Cl2溶液中,几种目标产物在620 nm左右均有较强的紫外吸收峰。BDP1和BDP2的最大吸收峰几乎处于同一波长(620 nm),且吸收峰轮廓也相似,而BDP3的最大吸收峰则相对更趋向于长波方向(649 nm),吸收峰轮廓也更宽阔。这主要是因为三苯胺拥有更强的给电子能力,其与BODIPY核之间的D-A效应更为强烈,引起的ICT也更为明显,降低了分子的能隙。BDP 1-3在溶液中均表现出良好的光谱吸收强度(6.8×104 -1.1×105M-1 cm-1),这可使分子高效利用光谱响应范围内的光子转换为光电流,具备良好的光伏电池材料特征。
相对于溶液中的光谱吸收,BDP 1-3在固态下的光谱吸收均表现出5-9 nm的红移,而且光谱吸收轮廓变得更为宽阔,吸收边界扩展到750-850 nm的区间。这是由于有机分子在薄膜中比在溶液中有更好的共面性,共轭效应更强,形成更加紧密的π-π堆积所致。
表2结果显示,几种染料的第一氧化电位分别是1.19、1.10和0.96 eV,计算得BDP1-3对应的HOMO能级分别为:-5.45,-5.37和 -5.23 eV;LUMO能级分别为:-3.45,-3.37和 -3.32 eV。可以看到这几个分子的HOMO能级均低于-5.2eV,因此几个分子在空气中具有较好的稳定性(-5.2eV是有机分子能够稳定存在的能级,低于这个数值分子在空气中能够稳定存在),适合于做有机小分子太阳能电池给体材料。
实施例2
中间体1的合成:
在2-碘-9,9’-二甲基芴 (3.2 g, 10 mmol)、双联频哪醇硼酯 (2.79 g, 11 mmol)、乙酸钾 (2.94 g, 30 mmol)、DMF (80 mL)的混合液中加入Pd(dppf)Cl2 (360 mg, 0.5 mmol)。体系在氩气保护下, 90oC反应8 h。停止反应,混合物冷却至室温后倒入200 mL 水中,用乙酸乙酯萃取 (50 mL × 3),再用饱和食盐水洗涤数次,合并有机相,无水硫酸镁干燥。减压旋转蒸发,去除溶剂,残余物以石油醚:乙酸乙酯 = 20:1 (v/v) 为洗脱剂用硅胶 (200-300目) 进行柱层析分离,得到白色固体2.82 g,产率88%。1H NMR (600 MHz, CDCl3) δ: 7.91 (s, 1H), 7.85 (d, J = 7.5 Hz, 1H), 7.76 (t, J = 6.8 Hz, 2H), 7.48-7.45 (m, 1H), 7.37-7.34 (m, 2H), 1.53 (s, 6H), 1.40 (s, 12H) 。
中间体2的合成:
在3-溴-9-辛基咔唑 (2 g, 5.6 mmol)、双联频哪醇硼酯 (1.7 g, 6.7 mmol)、乙酸钾 (2.7 g, 28 mmol)、DMF (80 mL)的混合液中加入Pd(dppf)Cl2 (150 mg, 0.21 mmol)。体系在氩气保护下, 90oC反应8 h。停止反应,混合物冷却至室温后倒入200 mL 水中,用二氯甲烷萃取 (50 mL × 3),再用饱和食盐水洗涤数次,合并有机相,无水硫酸镁干燥。减压旋转蒸发,去除溶剂,残余物以石油醚:乙酸乙酯 = 20:1 (v/v) 为洗脱剂用硅胶 (200-300目) 进行柱层析分离,得到白色固体1.7 g,产率76%。
中间体3的合成:
中间体3 的反应条件与合成中间体2的条件类似,用4-溴-N,N-二(4-异辛基苯基)苯胺作为反应底物。纯化后得到白色固体, 产率84%。
中间体4的合成:
在100 mL单口瓶中加入对辛氧基苯甲醛 (2.34 g, 10 mmol) 和新蒸的吡咯 (30 mL, 430 mmol), 向溶液中鼓氩气置换10 min,在氩气保护下迅速加入催化剂InCl3 (0.11 g, 0.5 mmol), 在室温下磁力搅拌5 h, 向其中加入NaOH (0.2 g, 5 mmol)粉末继续搅拌30 min,终止反应。减压蒸馏,回收多余的吡咯,得粗产品,用硅胶(200-300目)柱层析[洗脱液,V(石油醚):V(乙酸乙酯) = 7:1],纯化得到白色晶体2.45 g, 产率70%。
在500 mL三口瓶中,取上述合成的白色晶体 (3.50 g, 10 mmol) 溶解于二氯甲烷 (80 mL) 中,加入四氯苯醌 (2.9 g, 12 mmol), 室温条件下磁力搅拌,充分氧化8 h。然后将反应混合液置于氩气保护下,慢慢滴加三氟化硼乙醚 (37 mL, 300 mmol),搅拌反应10 min 后再缓慢加入三乙胺 (41.7 mL, 300 mmol),滴加完毕后继续反应8 h。混合物倒入氢氧化钠溶液中,用二氯甲烷萃取。有机相用无水硫酸钠干燥。过滤、滤液减压下去除溶剂。粗产品用硅胶 (200-300目) 柱层析[洗脱液,V(石油醚):V(乙酸乙酯)=10:1] 分离得红绿色粉末状化合物2.50 g,产率63%。
向100 mL三口瓶中依次加入上一步合成的红绿色固体 (1.0 g, 2.5 mmol)、DMF (20 mL) 和二氯甲烷 (20 mL),在室温下磁力搅拌几分钟后,装上恒压滴液漏斗,抽真空,通入氩气保护,将NBS (0.89 g, 5.0 mmol) 溶解在DMF (5 mL) 和二氯甲烷 (5 mL)中,通过恒压滴液漏斗逐滴滴入到三口瓶中,滴加完毕后再继续反应4 h,整个过程避光。向三口瓶中加入50 mL去离子水,用二氯甲烷萃取三次 (80 mL × 3),有机层再经饱和食盐水洗涤 (100 mL × 3),合并有机相,无水硫酸镁干燥过夜。过滤收集滤液,减压下旋转蒸发去除溶剂,得粗产品,用硅胶(200-300目)柱层析[洗脱液,V(石油醚):V(乙酸乙酯) = 20:1]纯化得红色粉末状固体 0.97 g,产率70%。
中间体5的合成:
在100 mL单口瓶中,依次加入中间体4 (0.55 g, 1.0 mmol)、2-(三丁基锡)噻吩(0.45 g, 1.2 mmol), 氟化铯 (0.3 g, 2.0 mmol) 和甲苯 (30 mL),抽真空,再向反应瓶中加入四(三苯基膦)钯 (0.15 g, 0.13 mmol),氩气保护,120oC下磁力搅拌反应16 h。停止反应,冷却至室温,倒入水中,用乙酸乙酯萃取,再用饱和食盐水洗涤 (30 mL × 3),收集有机相,无水硫酸钠干燥。减压下旋转蒸发去除溶剂,得粗产品,用硅胶 (300-400目) 柱层析 [洗脱液,V(石油醚):V(乙酸乙酯) = 10:1],得到褐色粉末状化合物固体0.31 g,产率56%。
BDP1的合成
在100 mL 单口瓶中,依次加入中间体1 (96 mg, 0.3 mmol) 、 中间体5 (140 mg, 0.25 mmol)、四(三苯基膦)钯 (11 mg, 0.01 mmol)、甲苯 (30 mL) 和碳酸钾溶液 (20 mL 2M),排除空气,氩气保护,混合物在分别在65℃、85℃、115℃下反应12h、24h、34h后停止反应。将反应混合液冷却至室温,倒入100 mL 蒸馏水中,用乙酸乙酯萃取数次,有机层再用饱和食盐水洗涤 (50 mL×3),合并有机相,无水硫酸镁干燥。减压下旋转蒸发去除溶剂。残余物以石油醚:乙酸乙酯 = 10: 1 (v/v)为洗脱剂用硅胶 (300-400目) 进行柱层析分离提纯,得到墨绿色固体204 mg,产率61%。
BDP2的合成
向100ml圆底烧瓶中加入中间体2和中间体5,反应条件和合成BDP1的条件相同,纯化得到墨绿色固体,产率63%。
BDP3的合成
向100ml圆底烧瓶中加入中间体3和中间体5,反应条件和合成BDP1的条件相同,纯化得到墨绿色固体,产率59%。
本发明通过上述实施例来说明本发明的详细合成方法,但本发明并不局限于上述方法,即不意味着本发明必须依赖上述反应条件才能实施。所属技术领域的技术人员应该明了,对本发明的任何改进,对本发明反应溶剂催化剂的等效替换及反应具体条件的改变等,均落在本发明的保护范围和公开范围之内。
- 还没有人留言评论。精彩留言会获得点赞!
- 一种二苯并噻吩的萃取方法与流程
- 取代的苯并噻吩基衍生物作为用于治疗II型糖尿病的GPR40激动剂的制造方法与工艺
- 2‑哌嗪‑1‑基‑4H‑1,3‑苯并噻嗪‑4‑酮衍生物及其用于治疗哺乳动物感染的用途的制造方法与工艺
- 一种甲硫基取代苯并[d]恶唑衍生物的制备方法与流程
- 包括一种或多种1,1‑二取代的烯烃化合物的乳液聚合物、乳液法及聚合物组合物的制造方法与工艺
- 包括一种或多种1,1‑二取代的烯烃化合物的聚合物及其聚合物组合物的制造方法与工艺
- 包括一种或多种1,1‑二取代的烯烃化合物的溶液聚合物、溶液聚合法及聚合物组合物的制造方法与工艺
- 一种异原子取代的苯并噻二唑基聚合物给体材料及其制备方法和应用与流程
- 5,7位取代的1H‑苯并[e][1,4]二氮杂‑2(3H)‑酮衍生物的制备方法与流程
- 具有两个二苯并呋喃或二苯并噻吩取代基的咔唑的制造方法与工艺