一种定点整合中国仓鼠卵巢细胞株的改造及其用途的制作方法
本发明涉及细胞基因工程领域,具体涉及一种定点整合外源基因的FRT-CHOS细胞系及其在蛋白表达的用途。
背景技术:
提高哺乳动物细胞外源基因表达水平和稳定性,一直是生物制药领域或基因工程领域不断发展的需求。尽管大多数商业化表达体系尤其表达载体都有针对性的优化设计,如选择强启动子,带有加压筛选系统等,但获得高表达外源蛋白的细胞仍然需要耗费巨大的精力和成本。通过常规技术引入外源基因的整合具有随机性,插入位置不支持强转录发生,并导致蛋白表达水平较低,即使短时的高水平表达,但也不能维持其稳定性,并随传代次数增加表达水平逐渐下降。因此,外源基因插入染色体的“位置效应”以及整合位点是否在高转录活跃区等被提出是影响蛋白表达水平及其稳定性的重要因素。只有对目的基因实施定向整合如整合至基因组的特定位置,才可能实现外源基因的稳定整合和蛋白高水平表达。
外源基因定向整合方法有多种,包括同源重组技术衍生的位点特异性重组技术,依靠针对特异识别位点的整合酶,在基因组和外源DNA间实现基因置换、基因敲出及敲入等基因工程操作。由于能够有效克服靶向性低、随机整合以及位置效应影响等缺点,定点整合技术在基因工程领域中的应用已为外源基因靶向定点整合奠定了良好基础。从20世纪80年代发展至今,位点特异性重组技术已有多种,其中最成熟的技术为FLP/FRT系统和Cre/loxP系统。FLP/FRT技术在哺乳动物表达系统中的应用开始于20世纪90年代初期。O’Gorman等应用FLP重组酶技术实现了将外源基因整合进入预先确定的哺乳动物细胞染色体位点(Science.1991Mar 15;251:1351-5.Recombinase-mediated gene activation and site-specific integration in mammalian cells);Wiberg FC等应用FLP/FRT技术将编码抗体的基因整合进入CHO细胞基因组,首先在CHO细胞基因组特定位点插入1个FRT位点,建立含有单个FLP重组酶识别位点的宿主细胞系,然后将同样含有单个FRT位点的抗体表达载体以及FLP重组酶表达载体共转染宿主细胞,编码抗体的基因在FLP重组酶的作用下,定点整合在细胞基因组的相同位点,有效克服了位置效应对抗体表达水平的影响(Biotechnol Bioeng.2006,94:396-405.Production of target-specific recombinant human polyclonal antibodies in mammalian cells.)。目前,通过FLP/FRT技术已推出系列商业化细胞系,包括Flp-InTM-293、Flp-InTM-CV-1、Flp-InTM-CHO、Flp-InTM-BHK和Flp-InTM-3T3等,这些细胞系均可借助重组酶体系完成细胞基因组与外源DNA的位点特异性重组。然而,所述细胞系均为贴瓶生长,作为产业化用途尤其在表达治疗性蛋白或治疗性单克隆抗体方面的运用具有很大的局限性。中国专利(申请号:200310115022.4)公开了一种CHO/dhfr-细胞定点整合表达系统,该系统能够实现外源基因在CHO/dhfr-细胞基因组中定点整合和有效表达。PCT专利(2012)公开了一种染色体着陆垫(chromosomal landing pad)及其相关用途,尤其公开了强转录活性位点位于宿主细胞的Ank2、Cpsf4、C-Mos、Nephrocystin-1/Mal基因之内或附近的染色体着陆垫,这些染色体“着陆垫”均借助FLP/FRT技术实现,并成功用于外源基因的定点整合。中国专利(申请号:201510130235.7)公开了一种CHO细胞定点整合表达系统,该系统能够实现外源基因在细胞染色体基因组中1q12及1q13区实现定点整合和高效表达。
作为产业化应用的中国仓鼠卵巢(CHO)细胞,具有多种亚型或衍生细胞系,如CHO-K1,CHO-DG44以及CHO-S细胞,这些细胞各自的染色体总数以及结构畸变不尽相同,但均已发生大的变异,多数染色体已经发生重排(Deaven LL,Petersen DF,Chromosoma.1973,41∶129-144The chromosomes of CHO,an aneuploid Chinese hamster cell line:G-band,C-band,and autoradiographic analyses;Derouazi M et al,Biochemical and Biophysical Research Communications.2006.340∶1069-1077.Genetic characterization of CHO production host CHO-DG44and derivative recombinant cell lines),以CHO-K1和CHO-DG44为例,仅有少数染色体保留了其正常结构,本发明人在前期研究的基础上,通过在CHO细胞实施电转染外源基因发现,除了整合至1号染色体1q13区的外源基因能高效稳定表达外,1号染色体的1p12区也可以实现高效表达。本发明由此设计在悬浮生长的CHO-S细胞基因组中插入FRT位点,从中筛选出整合在不同染色体上的单个FLP重组酶识别位点的细胞,其能够实现外源蛋白的高效表达,由此产生的本发明可接受任何含有同样单个FRT位点的蛋白或抗体表达载体,并用于后续产业化表达。
技术实现要素:
本发明的目的是提供一种基因工程FRT-CHOS细胞系及其用途,包括:
(1)悬浮培养的中国仓鼠卵巢(CHO)细胞系,优选CHO-S细胞系;
(2)应用FLP/FRT技术将含FRT识别标签载体通过电转染方式插入悬浮培养的CHO-S细胞基因组;
(3)逐步提高博来霉素浓度加压选择Zeocin抗性克隆,获得FRT随机整合的混合细胞;
(4)经有限稀释进行细胞亚克隆,结合荧光原位杂交技术筛选单个稳定整合FRT位点在1p12区的FRT-CHOS55细胞作为本发明FRT-CHOS细胞;
(5)应用Flp重组酶技术,在细胞内介导使外源基因插入本发明FRT-CHOS55细胞FRT识别位点,通过共转染表达Flp重组酶的质粒以及表达目的蛋白的质粒pFRT-GIA,使Flp重组酶在瞬时表达的同时,促使pFRT-GIA特异性在FRT位点实施同源重组,实现目的基因的定点整合。
附图说明
图1 UCOE-dhfr质粒结构示意图
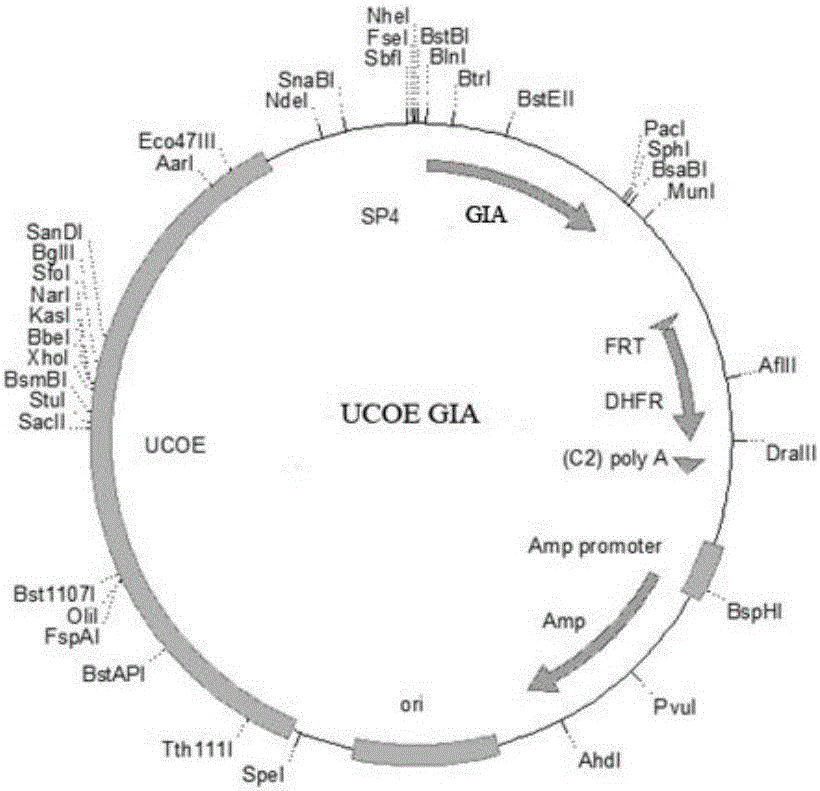
图2 UCOE-GIA质粒结构示意图
图3 pBAT-FIp质粒结构示意图
图4 工程细胞系FRT-CHOS55定点整合的荧光原位杂交鉴定
图5 工程细胞系外源蛋白表达水平比较(D:天)
图6 工程细胞系外源蛋白表达稳定性比较(p:代次)
具体实施方式
提供以下实施例对本发明作进一步阐释。应当理解,这些实施例仅用于说明本发明而非对本发明有任何限制。
实施例1FRT识别位点在CHO细胞染色体的定位筛选
1、细胞培养
CHO-S细胞系,在CD FortiCHOTM Medium培养基中添加谷氨酰胺及抗结团剂(Anti-Clumping Agent),在5%CO2,37℃,130rpm/min的细胞摇床中进行培养。
2、质粒转染及细胞初步筛选
参照无内毒素质粒试剂盒说明书(DP117,天根生化科技有限公司)提取质粒pFRT/lacZeo,利用ScaI限制性内切酶将pFRT/lacZeo质粒进行线性化处理,酶切产物回收后,采用Lonza Cell Line KitV试剂盒并参照说明书(VCA-1003,LONZA)进行pFRT/lacZeo质粒电转染。转染质粒后的细胞置于摇瓶培养至48h,细胞计数仪检测细胞活率和细胞数。转染后进行逐级加压筛选:博来霉素(Zeocin)浓度为50μg/mL,100μg/mL,250μg/mL,500μg/mL,750μg/mL和1000μg/mL,逐渐加压,获得初筛细胞。
3、单克隆细胞筛选
将初筛细胞转入半固体培养基,半固体接种方法步骤同前,挑取获得的单克隆细胞系通过检测转染细胞中lacZ基因产物β-半乳糖苷酶活性进行初步筛选,β-半乳糖苷酶活性检测方法参照试剂盒(β-Gal Assay Kit,K1455-01,Thermo)提供的说明进行。经连续2次半固体培养基筛选,挑取数个单克隆细胞系,根据β-半乳糖苷酶活性高低进行筛选的单克隆细胞系;并通过对单克隆细胞系提取基因组DNA,利用特异引物LacZF和LacZR通过PCR扩增目的基因,确认目的载体整合至基因组中。细胞系用于后续研究。
4、单克隆细胞染色体荧光原位杂交
a)染色体制片
优选15个高表达β-半乳糖苷酶活性的单克隆细胞系分别扩大培养,加入秋水仙素(终浓度为0.5μg/m1)孵育2h;取15ml离心管收集细胞,1000rpm离心5min,去上清;用37℃预热的KC1(0.075mol/L)溶液1ml重悬细胞,补加7ml KC1(0.075mol/L)溶液,37℃水浴低渗处理20min;加入2ml固定液(甲醇:冰乙醇(v∶v)=3∶1),轻轻吹打混匀后放置10min,1000rpm离心10min,去上清;加入1 mL固定液重悬细胞,加3mL固定液混匀,室温放置10min,200g离心10min,去上清留约0.5mL体积;重悬细胞。取预冷的玻片,用玻璃吸管滴片(每片滴加40-50μL的细胞悬液),室温放置24h。
b)探针制备
取20μg pFRT/lacZeo质粒,利用HindIII和Bpu10I限制性内切酶进行双酶切处理,37℃条件反应过夜,切胶回收小目的片段,用于探针标记模板,该小片段与整合的质粒互补,将小片段利用非放射性生物素进行标记,具体标记操作步骤按照Random Primed DNA Labeling Kit试剂盒说明书进行,获得FRT探针。
c)荧光原位杂交
将室温放置24h的玻片分别在变性液(2×SSC/70%甲酰胺)中75℃变性2min,依次置入70%、90%、100%冰乙醇中各2-5min脱水,室温干燥;探针用杂交液稀释至终浓度10ng/μL,78℃变性8min,37℃复性45min,加在变性处理后的样本玻片上,封片,37℃湿盒中杂交过夜;去除封片,43℃水浴,2×SSC/50%甲酰胺15min;用2×SSC/0.01%Tween 20洗片三次,室温5min,2×SSC洗片两次,室温5min;玻片室温晾干后,暗室滴加30μL链霉亲和素-FITC,盖上盖玻片,37℃暗处温浴1h,去除盖玻片。用2×SSC/0.01%Tween 20洗片三次,室温5min,2×SSC洗片两次,室温5min;依次置入70%、90%、100%冰乙醇中各5min脱水,暗处室温挥干;加入自配含DAPI的抗淬灭液复染,覆盖玻片(乙醇预先处理),10min后在荧光显微镜下观察。
原位杂交分析结果显示,外源标签随机插入CHO细胞基因组中,共计获得14个整合位点不同的单克隆细胞系(见表1),选择其中1株β-Gal活性最高的细胞系和1株活性最低的对照细胞株用于同源重组,分别为FRT插入位点在1p12的FRT-CHOS55细胞系以及FRT插入位点在Z8的FRT-CHOS6细胞系。
表1 FRT外源标签插入位点
实施例2外源基因同源重组及蛋白表达
1、表达质粒构建
a)UCOE-dhfr表达载体构建
UCOE-dhfr载体通过人工改造获得,其可用于外源基因定点整合及蛋白表达。利用特异引物P1和P2通过PCR方法扩增获得基因片段。PCR反应体系1(总体积50μl):5×Buffer 10μl、dNTP 2μl、引物1μl(P1和P2)、模板为UCOE载体1μl、Phusion酶0.5μl,最后用双蒸水补至50μl;反应条件:98℃预变性60s,1个循环,98℃10s、55℃30s、72℃50s,30个循环,最后72℃7min。基因产物以琼脂糖凝胶电泳检测,获得基因片段命名为genel。利用特异引物P3和P4通过PCR方法扩增获得基因片段DHFR。PCR反应体系1(总体积50μ1):5×Buffer 10μl、dNTP 2μl、引物1μl(P1和P2)、模板为pCHO1.0载体1μl、Phusion酶0.5μl,最后用双蒸水补至50μl;反应条件:98℃预变性60s,1个循环,98℃10s、55℃30s、72℃50s,30个循环,最后72℃7min。基因产物以琼脂糖凝胶电泳检测,获得基因片段命名为gene2。利用特异引物P1和P4通过拼接PCR方法扩增获得基因片段。PCR反应体系1(总体积50μl):5×Buffer 10μl、dNTP 2μl、引物1μl(P1和P2)、模板为pCHO1.0载体1μl、Phusion酶0.5μl,最后用双蒸水补至50μl;反应条件:98℃预变性60s,1个循环,98℃10s、55℃30s、72℃50s,30个循环,最后72℃7min。基因产物以琼脂糖凝胶电泳检测,获得基因片段命名为gene3。Gene3包含了mPKG启动子、FRT位点和DHFR基因序列。将gene3和UCOE载体分别利用SalI和DraIII双酶切处理,前者利用产物回收试剂盒进行基因片段回收,后者利用胶回收试剂盒回收载体大片段,连接,构建UCOE-dhfr载体,转化Top10大肠杆菌感受态细胞,挑取单克隆,提取质粒酶切及序列分析,结果UCOE-dhfr载体构建成功(图1)。
引物如下所示:
P1:5-AGAGTCGACCCTAGGGATATCTTAATTAACTGCAGGCATGCAAGCTGGC-3
P2:5-CTTCGGAATAGGAACTTCGGTAAGCTGGTCGAAAGGCC-3
P3:5-GAAGTTCCTATTCCGAAGTTCCTATTCTCTAGAAAGTATAGGAACTTCATGGTTCGACCATTG-3
P4:5-ATGCACCAGGTGTTAGTCTTTCTTCTCGTAGAC-3
b)UCOE-GIA质粒构建
UCOE-GIA载体通过人工改造获得,其在UCOE-dhfr载体中插入外源目的基因,用于外源基因定点整合及蛋白表达。全基因合成方法获得目的基因片段,获得基因片段命名为GIA。UCOE-dhfr和GIA分别进过AvrII和PacI酶切处理。在T4连接酶的作用下,将基因片段UCOE-dhfr、GIA基因片段连接。连接反应体系如下:在1.5ml EP管中加入如下成分UCOE-dhfr0.5μl,GIA 7.5μl,10×T4Buffer1μl,T4DNA ligase1μl,混匀后在室温(20℃左右)下反应4h以上,连接产物转化至Top10大肠杆菌感受态细胞中,涂布于2YT平板培养基上37℃静止过夜,平板编号UCOE-GIA。从UCOE-GIA平板中各挑取数个重组单菌落经过培养后做为PCR模板,分别进行PCR筛选鉴定。菌液PCR扩增反应体系(总体积20μL):2×Taq HS 10μL、菌液模板2μL、上下游引物各1μL(终浓度0.3μmol/L),最后用双蒸水补至20μL;反应条件:95℃2min,一个循环;94℃60s、53℃60s、72℃120s,30个循环;最后72℃5min。通过菌落PCR鉴定正确的菌落接种后提取再进行酶切鉴定。首先进行重组菌的质粒提取,然后进行酶切分析,将通过菌落PCR、酶切鉴定及测序鉴定获得正确UCOE-GIA质粒(图2)。
2、pBAT-Flp质粒构建
pBAT-Flp载体通过人工改造获得,用于特异性表达FLP重组酶。利用特异引物P5和P6通过PCR方法扩增获得基因片段。PCR反应体系(总体积50μl):5×Buffer 10μl、dNTP 2μl、引物1μl(P1和P2)、模板为pOG44载体1μl、Phusion酶0.5μl,最后用双蒸水补至50μl;反应条件:98℃预变性60s,1个循环,98℃10s、55℃30s、72℃50s,30个循环,最后72℃7min。基因产物以琼脂糖凝胶电泳检测,获得基因片段命名为Flp。将Flp和pCI-neo载体分别利用NheI和SalI双酶切处理,前者利用产物回收试剂盒进行基因片段回收,后者利用胶回收试剂盒回收载体大片段,连接,构建pBAT-Flp载体,转化Top10大肠杆菌感受态细胞,挑取单克隆,提取质粒酶切及序列分析,结果pBAT-Flp载体构建成功(图3)。
引物如下所示:
P5:5-GCCGCTAGCATGCCACAATTTGATATATTATG-3
P6:5-GCCGTCGACTTATATGCGTCTATTTATG-3
3、质粒共转染
质粒pBAT-Flp含有FLP重组酶表达元件,通过与特定质粒的共转染,可实现特定质粒在基因组中FRT位点的定点整合。采用已筛选的FRT整合工程细胞系FRT-CHOS55和FRT-CHOS6进行检测,按照Lonza Cell Line Kit V试剂盒说明书分别进行UCOE-GIA和pBAT-Flp质粒共转染。转入质粒的细胞分别置于摇瓶培养(37℃、5%CO2、130rpm/min)至48h,细胞计数仪检测细胞活率和细胞数。
转染48h后进行加压筛选:即MTX浓度从100nM起逐渐加至200nM、500nM、700nM、和1000nM,并获得初筛细胞,再经有限稀释法进行单克隆细胞筛选,获得单克隆细胞系FRT-CHOS55-GIA和FRT-CHOS6-GIA。
4、同源重组鉴定
a)荧光原位杂交
i.染色体制片
单克隆细胞系染色体制备方法参照实施例1中实验4a操作。
ii.探针制备
以UCOE-GIA质粒,利用AvrII和PacI限制性内切酶进行双酶切处理,37℃条件反应过夜,切胶回收小片段用于探针标记,该小片段与整合的质粒互补,将小片段利用非放射性生物素进行标记,具体标记操作步骤按照Random Primed DNA Labeling Kit试剂盒说明书进行,获得探针。
iii.荧光原位杂交
荧光原位杂交实验方法参照实施例1中实验4c操作。
原位杂交分析结果显示,FRT-CHOS55-GIA细胞系目的基因特异性整合至1号染色体1p12处,FRT-CHOS6-GIA细胞系目的基因特异性整合至Z8号染色体Z8p1处(图4),说明目的基因与细胞系的FRT位点实现了定点整合。
b)外源蛋白表达
筛选阳性克隆细胞从96孔板扩至24孔板生长,然后扩至6孔板生长,之后扩至50mL摇瓶生长。分别收集培养3天、5天、7天、9天、11天细胞培养上清液,离心除去细胞碎片,上清液进行蛋白鉴定与蛋白浓度检测。Western blot检测结果GIA蛋白分子量大小符合理论值,表明GIA蛋白正确表达。蛋白浓度检测结果显示,工程细胞系能够实现外源蛋白高水平表达,筛选获得的稳定转染FRT-CHOS55-GIA细胞系其11天细胞培养上清表达量达到1.23g/L。
同时对转染的2个细胞系(包括FRT插入位点在1p12的FRT-CHOS55-GIAD单克隆细胞系、FRT插入位点在Z8p1的FRT-CHOS6-GIAD单克隆细胞系)及其对应的pool细胞(包括FRT-CHOS55-GIA-pool细胞系、FRT-CHOS6-GIA-pool细胞系)的外源蛋白表达水平进行比较,分别收集培养3天、5天、7天、9天、11天细胞培养上清液,离心除去细胞碎片,上清液进行蛋白鉴定与蛋白浓度检测。蛋白浓度检测结果发现FRT-CHOS55细胞系能够实现外源蛋白高水平表达,而FRT-CHOS6工程细胞系其外源蛋白水平表达较低(图5)。
5、外源蛋白表达稳定性考察
单克隆细胞系FRT-CHOS55-GIAD和FRT-CHOS6-GIAD经过长期传代培养,分别考察了多代次的细胞分泌外源蛋白表达水平,通过对10代、20代、30代、50代、70代和100代细胞的外源蛋白表达水平进行检测,结果发现经过长期传代培养后,FRT-CHOS55-GIAD工程细胞系能够实现外源蛋白稳定表达,100代次的表达量与0代相比下降幅度为20%左右,说明单克隆细胞系FRT-CHOS55-GIAD外源蛋白表达稳定性较好,说明工程细胞系FRT-CHOS55可用于高效稳定表达外源蛋白的应用;而FRT-CHOS6-GIAD工程细胞系其外源蛋白表达稳定性较差,100代次的表达量与0代相比下降幅度接近40%(图6)。
- 还没有人留言评论。精彩留言会获得点赞!