一种普卡必利中间体的制备方法与流程
本发明属于医药合成领域,尤其涉及一种制备普卡必利中间体的方法。
背景技术:
普卡必利,化学名为 4-氨基-5-氯-2,3-二氢-N-[1-(3-甲氧基丙基)-4- 哌啶基]-7-苯并呋喃甲酰胺,临床上常用其琥珀酸盐,作为高选择性的 5-羟色胺受体激动剂。它能刺激人的肠蠕动反射,增强结肠收缩,加速胃排空。能增加便秘患者的排便频率,并降低粪便硬度,主要用于治疗各种便秘及手术后胃肠道蠕动迟缓和假性肠梗阻,是一种效果显著的肠动力药。临床上用于轻泻无法充分缓解的女性慢性便秘的对症治疗。普卡必利的上市给需要长期服药治疗慢性便秘而又担心因长期服用泻剂而引起各种不良反应的患者,提供了更安全、更有效、更为对症的治疗手段。
在众多普卡必利合成路线中,4-氨基-5-氯-2,3-二氢苯并呋喃-7-羧酸是必不可少的中间体之一。在现有的报道中,该中间体有如下三条合成路线:(1)Pharmaceutical and Clinical Research,2011,19(4),306-307.该路线第二步的重排反应温度近200℃,条件苛刻,共需要七步反应,合成路线偏长,其中第三步氧化反应用到四氧化锇,其价格比较高,所以该合成路线的合成成本较高;(2)J Heterocyclic Chem,1980, 17(6): 1333-5.该路线第二步和第六步反应均使用丁基锂,要求无水无氧低温操作,条件苛刻,工业化可行性差;(3)American Chemical Society,2003,125-139 . 该路线第三步反应为锌粉催化的合环反应,要求无氧操作,反应时有引发反应过程,易积聚放热,工业生产中存在安全隐患。
由此可见,现有的合成路线,均不易于工业化生产。亟需寻求一种原料易得,操作方便,反应条件温和的新的普卡必利中间体的制备方法。
技术实现要素:
本发明针对上述问题,提供一种新的普卡必利中间体的制备方法。该方法制备的中间体纯度高,收率高;合成路线简单易操作,安全,可行性强。
为了实现上述目的,本发明提供普卡必利中间体-氨基-5-氯-2,3-二氢苯并呋喃-7-羧酸的合成方法,合成路线如下。
所述的制备方法包括如下步骤:(1)以2-甲氧基-4-乙酰氨基-5-氯苯甲酸甲酯(Ⅰ)为起始原料,在适量的酰化试剂和路易斯酸作用下,控温0-20℃,在二氯甲烷溶剂中发生酰化及脱甲基反应,得到中间体(Ⅱ);(2)向得到的中间体(Ⅱ)中加入碱性试剂,在极性溶剂中控温0-20℃,发生合环反应,得到中间体(Ⅲ);(3)中间体(Ⅲ)中加入水合肼和无水乙醇,控温70-80℃,发生还原反应,得到中间体(Ⅳ);(4)中间体(Ⅳ)在氢氧化钠溶液作用下,控温90-100℃,发生水解反应,得到中间体(Ⅴ)的钠盐,进而用盐酸酸化,得到中间体(Ⅴ)。
所述的酰化试剂为氯乙酰氯或溴乙酰氯,优选氯乙酰氯。
所述的酰化试剂与2-甲氧基-4-乙酰氨基-5-氯苯甲酸甲酯的摩尔比为1.2:1。
所述步骤(1)中的温度优选0-10℃。
所述的路易斯酸为三氯化铝,其与2-甲氧基-4-乙酰氨基-5-氯苯甲酸甲酯重量比为1.2-1.8:1,优选1.5:1。
所述合环反应的碱性试剂为氢化钠、氢化钾、氢氧化钠、氢氧化钾、碳酸钾、碳酸钠、叔丁醇钠、叔丁醇钾、甲醇钠或乙醇钠中的任何一种,优选氢氧化钠。
所述合环反应的碱性试剂与中间体(Ⅱ)的摩尔比为1.5:1。
所述合环反应的极性溶剂为N,N-二甲基甲酰胺、甲酰胺、N,N-二甲基乙酰胺、二甲基亚砜、甲醇、乙醇或异丙醇中的任何一种,优选N,N-二甲基甲酰胺。
所述合环反应的温度优选为5-15℃。
本发明具备的有益效果。
本发明以2-甲氧基-4-乙酰氨基-5-氯苯甲酸甲酯为起始原料,生产厂家多,原料价格低、易得,其他辅助试剂也均为常用易得的化工原料;工艺路线中未采用易燃、易爆的丁基锂,以及毒性大、价格高的四氧化锇;采用工艺参数为0-100℃的温和反应操作,成功避开无水无氧的低温操作,避开200℃的高温操作,易于扩大工业化生产规模;工艺制备中,无锌粉使用,工艺合成路线中避免普卡必利中间体(Ⅴ)的脱氯杂质产生;普卡必利中间体(Ⅴ)的液相纯度高于99%,满足制备普卡必利必备中间体Ⅴ的质量要求;本合成工艺共四步反应,摩尔总收率达到37%,物料成本相比现有工艺降低近一半,适合工业化生产,能够为普卡必利的合成工艺提供经济适用的原料药。
附图说明
图1为中间体(Ⅴ)的 1H-NMR光谱图。
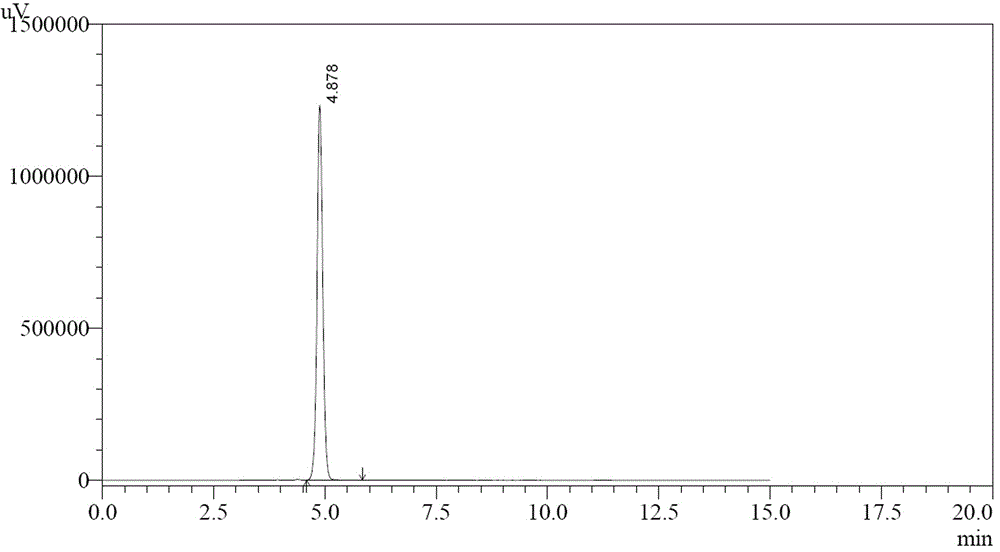
图2为中间体(Ⅴ)的液相色谱图。
具体实施方式
以下结合具体实施例对本发明技术方案作进一步详细说明。
实施例1。
将77 g三氯化铝加入到500mL二氯甲烷中,冷却至0-10℃,滴加26 g氯乙酰氯,控温0-10℃,搅拌0.5 h,搅拌转数300r/min。向反应体系中滴加起始物料Ⅰ的二氯甲烷溶液(50g+500mL二氯甲烷),温度为0-10℃,保温搅拌4h,搅拌转数300r/min;滴加500mL6%盐酸溶液,控制温度低于20℃,用分液漏斗分出有机层二氯甲烷,将有机层减压浓缩至无二氯甲烷蒸出(温度不高于60℃)得到油状物,油状物中加入50mL无水乙醇和450mL水,0-10℃打浆分散1h,抽滤,50-60℃鼓风干燥8h,得到中间体(Ⅱ),浅灰色固体52g,收率83%。
将50g中间体(Ⅱ)加入到300mL N,N-二甲基甲酰胺中,控温5-15℃,加入9.0g氢氧化钠,加毕,5-15℃搅拌16h,将反应体系倒入1500mL水中,析出固体,搅拌1h,搅拌转数300r/min,抽滤,50-60℃鼓风干燥8h,得到33g中间体(Ⅲ),收率76%。
将30g中间体(Ⅲ)加入到300mL无水乙醇中,升温至70-76℃,滴加30mL 80%水合肼,反应体系放热、放气,控制滴加速度保持体系微回流,加毕,70-80℃保温5h,加水500mL,常压蒸馏蒸出乙醇水溶液500mL,将体系冷却至10-20℃,析出固体,抽滤,50-60℃鼓风干燥8h,得到中间体(Ⅳ),浅棕色固体25g,收率89%。
将25g中间体(Ⅳ)加入到140mL水和27mL丙二醇单甲醚中,开动搅拌,加入20mL50%氢氧化钠水溶液,将体系升温至90-100℃,保温搅拌12h,冷却至0-10℃,搅拌1h,搅拌转数300r/min,抽滤得到固体(中间体Ⅴ的钠盐)。将中间体Ⅴ的钠盐加入到200mL水和40mL丙二醇单甲醚中,升温至75-85℃,滴加10mL浓盐酸,pH为1-2,析出固体,冷却至0-10℃,搅拌1h,搅拌转数300r/min,抽滤,水洗,50-60℃鼓风干燥8h,得到中间体(Ⅴ),白色固体13g,收率65%,纯度为100.0%(HPLC)。1H NMR (400 MHz, DMSO-d6) δ: 2.98 (t, J=8.8Hz, 2H), 4.60 (t, J=8.8Hz, 2H), 5.96 (s , 2H), 7.44 (s, 1H), 12.02 (s, 1H)。HPLC:100%,具体见图1-2。
实施例2。
将128g三氯化铝加入到1000mL二氯甲烷中,冷却至0-10℃,滴加52g氯乙酰氯,控温0-10℃,搅拌0.5h,搅拌转数300r/min。向反应体系中滴加起始物料Ⅰ的二氯甲烷溶液(100g +1000mL二氯甲烷),控温0-10℃,加毕保温搅拌4h。滴加1000mL6%盐酸溶液,控温低于20℃,用分液漏斗分出有机层二氯甲烷,将有机层减压浓缩至无二氯甲烷蒸出(温度不高于60℃)得到油状物,油状物中加入100mL乙醇和900mL水,0-10℃打浆分散1h,抽滤,干燥得到中间体(Ⅱ),浅灰色固体92g,收率73%。
将75g中间体(Ⅱ)加入到450mL N,N-二甲基甲酰胺中,控温5-15℃,加入37g碳酸钠,加毕,5-15℃搅拌16h,搅拌转数300r/min,将反应体系倒入3000mL水中,析出固体,搅拌1h,搅拌转数300r/min,抽滤,50-60℃鼓风干燥8h,得到60g中间体(Ⅲ),收率69%。
将30g中间体(Ⅲ)加入到300mL无水乙醇中,升温至70-76℃,滴加30mL80%水合肼,反应体系放热、放气,控制滴加速度保持体系微回流,加毕,70-80℃保温5h,加水500mL,常压蒸馏蒸出乙醇水溶液500mL,将体系冷至10-20℃,析出固体,抽滤,50-60℃鼓风干燥8h,得到中间体(Ⅳ),浅棕色固体26g,收率92%。
将25g中间体(Ⅳ)加入到140mL水和27mL丙二醇单甲醚中,开动搅拌,加入20mL 50%氢氧化钠水溶液,将体系升温至90-100℃,保温搅拌12h,搅拌转数300r/min,冷至0-10℃,搅拌1h,抽滤得到固体(中间体Ⅴ的钠盐)。将中间体Ⅴ的钠盐加入到200mL水和40mL丙二醇单甲醚中,升温至75-85℃,滴加10mL浓盐酸,pH为1-2,析出固体,冷却至0-10℃,搅拌1h,搅拌转数300r/min,抽滤,水洗,50-60℃鼓风干燥8h,得到中间体(Ⅴ),白色固体13.2g,收率66%,纯度为100.0%(HPLC)。1H NMR (400 MHz, DMSO-d6) δ: 2.98 (t, J=8.8Hz, 2H), 4.60 (t, J=8.8Hz, 2H), 5.96 (s , 2H), 7.44 (s, 1H), 12.02 (s, 1H),具体见图1-2。
需要指出的是,上述实施例仅为说明本发明的技术构思及特点,其目的在于让熟悉此项技术的人士能够了解本发明的内容并据以实施,并不能以此限制本发明的保护范围。凡根据本发明精神实质所作的等效变化或修饰,都应覆盖在本法的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!