催化效率提高的内切葡聚糖酶NfEG12A突变体及其编码基因和应用的制作方法
本发明涉及基因工程领域,具体地,本发明涉及催化效率提高的内切葡聚糖酶NfEG12A突变体及其编码基因和应用。
背景技术:
纤维素是自然界中含量最丰富的可再生能源之一,约占世界总生物质的50%左右。纤维素是由吡喃葡萄糖以不同比例的β-1,3和β-1,4糖苷键连接而成的长链多糖聚合物。β-葡聚糖链通过分子内和分子间氢键的紧密排列形成不可溶的纤维素微丝。
纤维素酶是指所有参与纤维素降解为葡萄糖的各种酶的总称。习惯上将纤维素酶分成三类:内切葡聚糖酶(endo-1,4-β-D-glucanase,EC3.2.1.4),其分子量大小约为23–146kDa,其随机定位于微纤维上的非结晶区,产生大量的还原端,催化域的活性中心为一个开放的裂隙;外切葡聚糖酶(exo-1,4-β-D-glucan cellobiohydrolase,CBH,EC3.2.1.91),其分子量约38–118kDa,结合于单根纤维素链的还原端或非还原端,持续性的降解结晶区的纤维素分子链;β-葡萄糖苷酶(β-glucosidase,EC3.2.1.21),其分子量约为40–250kDa,水解纤维寡糖或纤维二糖产生葡萄糖分子。在纤维素酶系中,三种酶相互协同催化纤维素酶的降解。
β-内切葡聚糖酶作为水解纤维素的关键酶,有着广泛的工业应用领域,如生物能源、纺织造纸、啤酒酿造、动物饲料添加及食品加工等。在饲料行业中添加β-葡聚糖酶可以帮助动物分解麦类成份,提高饲料中营养物质的利用率,促进动物的消化吸收和生长发育。在生物能源方面,随着石油能源的逐渐耗竭以及环境污染等严重问题,乙醇已被广泛的作为特殊的石油替代品,各国都在研究利用秸秆、稻壳、甘蔗渣、松子以及葵花籽等为原料提炼乙醇。提炼的过程中,纤维素酶和半纤维素酶可以将植物纤维降解为戊糖和己糖,以戊糖和己糖为发酵底物,利用微生物作用产生乙醇,而戊糖占木质纤维素含量的20–40%,因此可以大大降低生产成本。但是酶制剂的高成本及低效率问题一直制约着其在工业中的广泛应用,因此研究挖掘耐高温、耐酸碱及高比活的酶将具有更高的商业价值及竞争性。而自然界分离得到的酶很难满足工业需求,因此通过分子改良的方法,理性设计提高酶的性质是目前学术及产业界持续努力的目标。12家族内切葡聚糖酶NfEG12A是一种偏高温酸性酶,最适温度65℃,最适pH5.0,是目前已知的该家族中酶活最高的酶,为了进一步提高其工业应用潜能,我们对该酶进行分子工程改造,以提高其对纤维素降解的效率。本发明通过催化通道附近loop3的替换设计突变体来改良NfEG12A的催化效率,进而有效地提高其在工业上的应用价值。
技术实现要素:
本发明的目的是提供上述突变的内切葡聚糖酶。
本发明的再一目的是提供编码上述突变的内切葡聚糖酶的基因。
本发明的另一目的是提供包含上述基因的重组载体。
本发明的另一目的是提供包含上述基因的重组菌株。
本发明的另一目的是提供一种制备上述突变的内切葡聚糖酶的基因工程方法。
根据本发明的技术方案,延长了NfEG12A催化通道处loop3获得了催化效率提高的内切葡聚糖酶。
根据本发明的具体实施方式,延长了NfEG12A催化通道处loop3方式包括插入1、2、3、4或5个氨基酸残基。
本发明的另一目的提供上述突变的内切葡聚糖酶NfEG12FN和NfEG12STTQA的应用。
根据本发明的具体实施方式,本发明在氨基酸序列SEQ ID NO.1所示的内切葡聚糖酶NfEG12A中插入FN和STTQA序列,如SEQ ID NO.2和3所示,获得了催化效率提高的内切葡聚糖酶。
根据本发明的具体实施方式,设计各突变引物,以编码内切葡聚糖酶NfEG12A基因的野生型质粒pPIC9γ-NfEG12A为模板进行overlap PCR扩增。纯化得到含有突变体密码子的质粒,并将其转化进毕赤酵母感受态细胞GS115中进行表达,得到重组突变体蛋白。
根据本发明的具体实施方式,本发明对氨基酸序列SEQ ID NO.1所示的内切葡聚糖酶NfEG12A中插入FN序列,获得突变体NfEG12FN,其氨基酸序列如SEQ ID NO.2所示。
根据本发明的具体实施方式,本发明在氨基酸序列SEQ ID NO.1所示的内切葡聚糖酶NfEG12A中插入STTQA序列,获得突变体NfEG12STTQA,其氨基酸序列如SEQ ID NO.3所示。
内切葡聚糖酶NfEG12A氨基酸序列如SEQ ID NO.1所示:
MKTFAILGAFFSSALAQTLCDQYATYSNGRYTVNNNLWGKSSGSGSQCTYVDSISNSGVAWHTTWTWSGGDNQVKSYANSQVSLTKKLVSQISSIPTTVQWSYDNTNTRADVAYDLFTAADINHVTYSGDYELMIWLARYGSVQPIGSQIDSVNIGGHTWELWYGGSTQKTYSFVSATPITSFSGDVMDFWDYLTSRHGYPASSQYLINMQFGTEPFTGGPATLRVSQWTASVN
内切葡聚糖酶NfEG12FN氨基酸序列如SEQ ID NO.2所示:
MKTFAILGAFFSSALAQTLCDQYATYSNGRYTVNNNLWGKSSGSGSQCTYVDSISNSGVAWHTTWTWSGGDNQVKSYANSQVSLTKKLVSQISSIPTTVQWSYDNTNTRADVAYDLFTAADINHVTYSGDYELMIWLARYGSVQPIGSQIDSVNIGGHTWELWYGFNGSTQKTYSFVSATPITSFSGDVMDFWDYLTSRHGYPASSQYLINMQFGTEPFTGGPATLRVSQWTASVN
内切葡聚糖酶NfEG12STTQA氨基酸序列如SEQ ID NO.3所示:
MKTFAILGAFFSSALAQTLCDQYATYSNGRYTVNNNLWGKSSGSGSQCTYVDSISNSGVAWHTTWTWSGGDNQVKSYANSQVSLTKKLVSQISSIPTTVQWSYDNTNTRADVAYDLFTAADINHVTYSGDYELMIWLARYGSVQPIGSQIDSVNIGGHTWELWYGSTTQAGSTQKTYSFVSATPITSFSGDVMDFWDYLTSRHGYPASSQYLINMQFGTEPFTGGPATLRVSQWTASVN
本发明通过PCR的方式引入突变,获得突变体内切葡聚糖酶以及其编码基因。本发明还提供了包含上述内切葡聚糖酶突变体编码基因NfEG12FN或NfEG12STTQA的重组载体,命名为pPIC9γ-NfEG12FN或pPIC9γ-NfEG12STTQA。
本发明还提供了包含上述内切葡聚糖酶突变体编码基因NfEG12FN或NfEG12STTQA的重组菌株。
本发明还提供了一种制备催化效率提高的内切葡聚糖酶突变体的方法,包括以下步骤:
1)用上述的重组载体转化宿主细胞,得重组菌株;
2)培养重组菌株,诱导重组纤维素酶表达;
3)回收并纯化所表达的内切葡聚糖酶突变体。
本发明还提供了上述内切葡聚糖酶突变体的应用。
根据本发明中设计的突变体NfEG12FN和NfEG12STTQA对大麦葡聚糖和纤维素的催化效率有0.3和0.9倍的提升,且具有更好的耐热性和pH耐受性,因此更适于工业中的应用。
附图说明
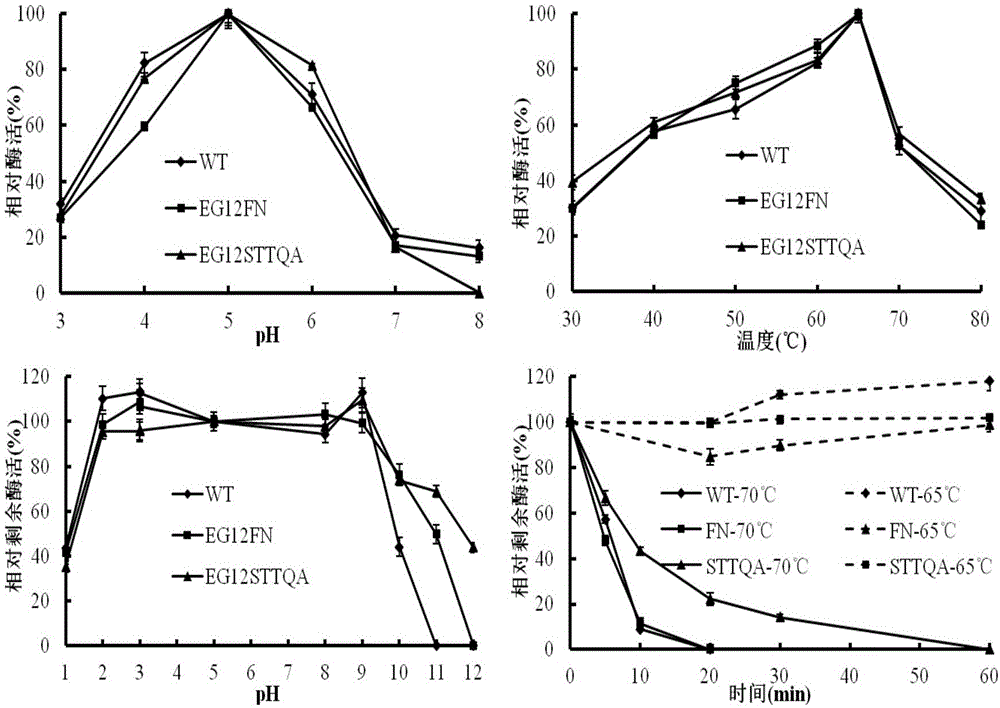
图1显示NfEG12A及其突变体的酶学性质测定。
图2显示NfEG12A及其突变体降解葡聚糖底物的酶活比较。
具体实施方式
试验材料和试剂
1、菌株及载体:毕赤酵母表达载体pPIC9及菌株GS115购自于Invitrogen公司。
2、酶类及其它生化试剂:内切酶购自TaKaRa公司,连接酶购自Sigma公司,其它都为国产试剂(均可从普通生化试剂公司购买得到)。
3、培养基:
(1)Neosartorya fischeri P1.培养基为马铃薯培养基(1000ml):200g土豆煮汁,10g葡萄糖,25g琼脂,pH5.0。
(2)大肠杆菌培养基LB(1%蛋白胨、0.5%酵母提取物、1%NaCl,pH7.0)。
(3)BMGY培养基:1%酵母提取物,2%蛋白胨,1.34%YNB,0.00004%Biotin,1%甘油(W/V)。
(4)BMMY培养基:除以0.5%甲醇代替甘油,其余成份均与BMGY相同,pH6.0。
说明:以下实施例中未作具体说明的分子生物学实验方法,均参照《分子克隆实验指南》(第三版)J.萨姆布鲁克一书中所列的具体方法进行,或者按照试剂盒和产品说明书进行。
实施例1构建突变体
一、克隆
以Neosartorya fischeri P1的cDNA为模板,以引物EG12-F和EG12-R对NfEG12A基因进行扩增。
电泳纯化PCR产物中的目的条带,用EorR I和Not I将PCR产物和pPIC9γ质粒进行双酶切,将酶切后的PCR产物和质粒用T4连接酶连接后得到pPIC9γ-NfEG12A重组质粒。
二、突变
突变体NfEG12FN和NfEG12STTQA是通过overlap PCR扩增构建的,其方法如下:
第一轮PCR是以pPIC9γ-NfEG12A质粒为模板,分别以EG12-F/NfEG12FN-R,NfEG12FN-F/EG12-R,EG12-F/NfEG12STTQA-R,NfEG12STTQA-F/EG12-R为引物进行PCR。
电泳回收第一轮PCR产物,分别命名为FN1,FN2,STTQA1,STTQA2。
第二轮PCR以第一轮PCR产物为模板,以EG12-F/EG12-R为引物进行扩增,电泳回收第二轮PCR产物。
电泳回收第二轮PCR产物,即得突变体片段NfEG12FN和NfEG12STTQA,用EorR I和Not I将PCR产物和pPIC9γ质粒进行双酶切,将酶切后的PCR产物和质粒用T4连接酶连接后得到pPIC9γ-NfEG12FN,pPIC9γ-eg12STTQA重组质粒。
所述构建pPIC9γ-NfEG12A的引物是:
EG12-F:5'-GCCGAATTCCAAACTCTCTGTGACCAGTATGCCAC-3'(SEQ NO.4)
EG12-R:5'-GCCGCGGCCGCCTAGTTCACACTGGCGGTCCACTGCG-3'(SEQ NO.5)
所设计的用于构建突变体的引物为:
NfEG12FN-F:
5'-GAGCTGTGGTACGGCTTCAACGGCAGTACCCAGA-3'(SEQ NO.6)
NfEG12FN-R:
5'-GTTGAAGCCGTACCACAGCTCCCAGGTATGGCC-3'(SEQ NO.7)
NfEG12STTQA-F:
5'-GAGCTGTGGTACGGCAGCACCACCCAGGCCGGCAGTACCCAGAAG-3'(SEQ NO.8)
NfEG12STTQA-R:
5'-GGCCTGGGTGGTGCTGCCGTACCACAGCTCCCAGGTATGGCC-3'(SEQ NO.9)
3、各突变体载体转化毕赤酵母GS115及工程菌的筛选
3.1制备受体毕赤酵母GS115感受态细胞;
3.2电击转化,以上述重组质粒转化感受态细胞。
3.3酵母转化子的筛选,筛选出具有多聚半乳糖醛酸酶活性的转化子;
3.4目的基因在毕赤酵母中摇瓶水平的表达:将含有较高的内切葡聚糖酶酶活菌株接种于300mL BMGY培养基的1L三角瓶中,置于30℃,220rpm摇床培养48h;后将培养液3000g离心5min,弃上清,沉淀用100mL含有0.5%甲醇的BMMY培养基重悬,并再次置于30℃,220rpm条件下诱导培养。每隔12h补加0.5mL甲醇,使菌液中的甲醇浓度保持在0.5%,同时取上清用于酶活性检测。
4、突变内切葡糖酶活力的测定
4.1内切葡聚糖酶的活性单位(U)定义:在给定的条件下,每分钟分解葡聚糖生成1μmolD-(+)-还原糖所需的酶量。
4.2重组酶反应最适pH和pH稳定性的测定:
将纯化好的重组内切葡聚糖酶在65℃下,不同pH的底物中进行酶学反应,以测定其最适pH。
pH稳定性测定:将浓缩后的纯酶液在不同pH值的缓冲液中置于37℃下处理1h,再用最适pH的缓冲液作适当稀释,在最适pH和最适温度的条件下测定剩余酶活性。
4.3重组酶反应最适温度和温度稳定性的测定:
葡聚糖酶的最适温度的测定为在柠檬酸-磷酸氢二钠缓冲液体系(pH5.0)及不同温度下进行酶促反应。耐热性测定为葡聚糖酶在65℃和70℃下分别处理10、20、30、60min后,于最适温度下进行剩余酶活性测定。
4.4重组酶比活、Km值及Vmax的测定重组酶
确定测定Km及Vmax的反应时间为5min。用不同浓度的葡萄糖(0.5,0.4,0.25,0.2,0.2,0.175,0.15、0.1、0.05%)为底物,在最适条件下测定酶活性,计算出相应的反应速度,利用GraphPad Prism 5软件计算Km值及Vmax。
按照Bio-Rad试剂盒的方法,绘制标准曲线。测定方法:首先通过标准曲线计算目的蛋白的含量,其次是在最适条件下测得重组酶的酶活,用酶活数除以蛋白的浓度可得酶的比活。比活力的定义为:每毫克酶蛋白所具有的酶活力单位数。
实验结果:
野生酶与突变酶的基本性质测定,结果如下:
如图1所述,与野生酶相比突变酶的最适温度与最适pH均没有改变,均为65℃,pH5.0。
在pH稳定性方面,野生酶的稳定范围在pH2~9,在pH1和pH10的条件下剩余约40%左右的相对酶活力。而突变体NfEG12FN在pH10和pH11还能剩余80%和60%的相对酶活力,突变体NfEG12STTQA在pH11和pH12还能剩余70%和50%的相对酶活力。
在热稳定性方面,野生酶与突变酶均在65℃下保持稳定,野生酶在70℃处理20min已丧失酶活力,而突变体NfEG12STTQA在70℃处理30min还剩余20%左右的相对酶活力。由此可以看出,突变体在pH稳定性和温度稳定性方面优于野生酶。
一、野生酶及突变酶的动力学实验测定
表1 NfEG12A及其突变体的动力学参数测定
与野生酶相比,两个突变体对地衣多糖和羧甲基纤维素钠的比活和催化效率均提高,其中NfEG12FN分别提高了近0.2和0.5倍,NfEG12STTQA分别提高了近0.8和1.3倍。寡糖催化效率分析发现,突变体对纤维六糖的催化效率也提高了,NfEG12FN提高了0.3倍,NfEG12STTQA提高了2.3倍。由此可以看出,突变酶的催化效率较野生酶有了比较明显的提高,这有利于其在工业中的应用。
二、野生酶与突变酶的底物特异性测定
表2 NfEG12A及其突变体的底物特异性测定
如图2所示,从结果可以看出,NfEG12A及其突变体均对混合键形成的β-葡聚糖表现出最强的水解能力,其中降解大麦葡聚糖的能力最强,地衣多糖次之;对β-1,4键形成的葡聚糖分子也有较强的水解能力;对支链型的多糖分子,如木葡聚糖和葡甘露聚糖具有微弱的降解能力,而对β-1,3键形成的昆布多糖分子及木聚糖没有降解能力。
- 还没有人留言评论。精彩留言会获得点赞!