减少引物二聚体扩增的方法与流程
本发明涉及在多重聚合酶链式反应(PCR)中减少引物二聚体扩增的方法。
发明背景
多重PCR由单个PCR混合物中的多个引物组组成,从而生成对不同DNA序列特异的扩增子。通过一次性靶向多个基因,可由单个测试轮次获得额外信息,否则需要若干倍的试剂和努力才能完成。
降低多重PCR测定灵敏度的主要障碍之一是引物二聚体(PD)的累积。PD由彼此杂交的引物分子组成,杂交的原因是成串的互补碱基,特别是在引物的3’端。多重PCR反应中极高浓度下许多引物对的存在提高了引物二聚体形成的机会。一旦形成,短的PD倾向于非常有效地扩增,通过大量消耗引物和其他试剂潜在地抑制所需DNA序列的扩增。可通过不同方法的组合来减少PD形成,包括特别的引物设计和修饰方法、使用热启动Taq聚合酶、PCR添加剂和优化的PCR循环条件。
已报道多种用于减少PD形成的引物设计和修饰方法。Brownie等(Nucleic Acids Res,25(16):3235-41,1997)描述了HANDS(同源标签辅助的无二聚体系统)。在HANDS PCR中,所有靶标特异性引物都在低浓度下在其5’端含有共有的尾部序列并在较高浓度下与单个尾部特异性引物混合。在至少两个循环的靶标特异性PCR后,对完全由尾部特异性引物驱动的后续扩增循环提高退火温度。结果是,来自所有PCR产物的单链(包括所需的扩增子和副产物如PD)都具有互补的5’和3’端,其导致形成相同的茎环结构。由于尾部序列的高局部浓度,短产物如PD中形成的茎环结构非常稳定且稳定程度超过尾部特异性引物的后续退火,导致PD扩增的抑制。然而,在所有引物各端均具有相同尾部序列的情况下,该方法需要靶扩增子足够的长以最小化真实靶产物上茎环的抑制作用。取决于高度多重PCR中靶扩增子的长度和组成,各扩增子的茎环的紧密度也发生变化,其可导致显著失衡的扩增。此外,该茎环可能没有足够稳定到抑制长引物之间的PD形成。
美国专利号5,792,607(Backman等)和美国专利申请公开号20140329245公开了使用内切酶IV切除引物的不可延伸3’,从而在特异性引物-模板杂交后激活引物。Dobosy等(BMC Biotechnol.11:80,2011)报道了RNA酶H依赖性PCR(rhPCR)方法,其使用RNA酶H切除位置靠近阻断的引物的3’端的单个RNA碱基,从而在特异性引物-模板杂交后激活引物。该方法最近由IDT(集成DNA技术公司(Integrated DNA Technologies),美国专利申请公开号2009/0325169,PCT/US2012/030413)商业化。所有这些方法都需要引物中修饰的碱基和用于引物激活的额外酶,这导致较高的成本。
Peleg等(Appl.Environ.Microbiol.,75:6393–6398,2009;WO/2009/004630)报道了PCR中的DNA-RNA嵌合引物减少PD形成。双启动寡核苷酸(DPO)引物(希捷科技(Seegene Technologies))已被报道用以减少PCR PD形成(Chun等,Nucleic Acids Res.35(6):e40,2007)。DPO包含两个单独的启动区域(5’端稳定子(stabilizer)和3’端决定子(determiner)),其通过聚脱氧肌苷接头接合。由于存在包含聚脱氧肌苷接头的弱氢键的“气泡”样结构,DPO引物3’端处的诸如PD的引物非特异性杂交减少。嵌合引物中的上述RNA碱基和DPO引物中的聚脱氧肌苷接头显著增加了引物制造的复杂程度和成本。
Scatterfield(J.Mol.Diagn.,16:163-173,2013)报道了由两条DNA序列组成的合作引物(cooperative primer),这两条DNA序列通过5’至5’或5’至3’的聚乙二醇接头接合。结果显示使用合作引物的单重PCR反应在添加的引物二聚体存在的情况下显著减少了引物-引物增殖。
尽管有这些努力,PD形成仍是多重PCR中的大挑战。具体而言,下一代测序(NGS)应用中靶标富集的多重程度极高,此时同一PCR反应池中存在数百甚至数千种引物。所有这些引物都可潜在地形成引物二聚体。
附图简要说明
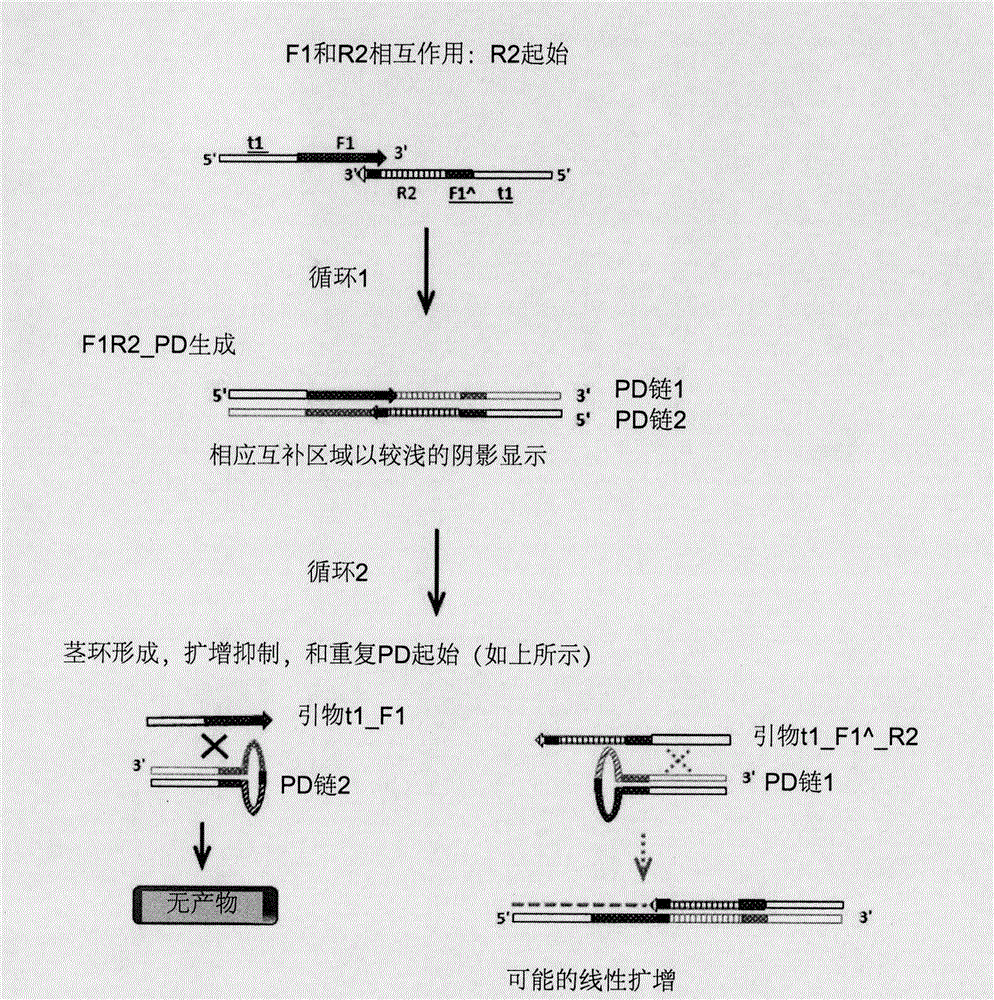
图1A显示用于扩增两条靶序列的PCR的第一循环和第二循环,其中t1F1、t2R1、t3F2和t1F1^R2为引物。图1B显示F1和R2的相互作用,以及引物二聚体的形成、扩增和抑制。
图2显示F1和R2在其3’端具有7个互补碱基且形成引物二聚体。
图3显示1阶段PCR扩增(顶部小图)和2阶段PCR扩增(底部小图)的PCR产物的凝胶电泳结果。泳道1:1重扩增子1,泳道2:1重扩增子2,泳道3-8:使用引物t1_F1^x_R2的2重,其中x分别为0、3、6、9、12和15个核苷酸。泳道M:50碱基DNA梯状条带。
发明详述
定义
“扩增子”指一段DNA或RNA,其是天然或人工的扩增或复制事件的来源和/或产物。在该文义中,“扩增”指遗传片段或靶序列的一个或多个拷贝(具体而言是扩增子)的生成。作为扩增反应的产物,扩增子可与诸如PCR产物的常用实验室术语互换使用。
“引物二聚体”(PD)是PCR的潜在副产物。PD由因引物中互补碱基而彼此杂交的引物分子组成。
本发明涉及一种减少多重聚合酶链式反应(PCR)中引物二聚体扩增的方法。当第一正向引物(F1)和第二反向引物(R2)在其3’端具有互补区域时,可能发生引物二聚体的形成。由于引物的高浓度,该互补区域可以是短至2-3个核苷酸以导致引物二聚体扩增。当该互补区域是至少4或5个核苷酸时,不需要的引物二聚体扩增几乎必然会发生。
本发明的方法在第一正向引物(F1)和第二反向引物(R2)在其3’端具有互补区域时减少引物二聚体问题。该方法包括以下步骤:(a)获得第一核酸序列,其包含第一标签(t1)和与第一靶核酸片段互补的第一正向引物(F1),(b)获得第二核酸序列,其包含第二标签(t2)和与第一靶核酸片段互补的第一反向引物(R1),(c)获得第三核酸序列,其包含第三标签(t3)和与第二靶核酸片段互补的第二正向引物(F2),(d)获得第四核酸序列,其包含第一标签(t1)、与第二靶核酸片段互补的第二反向引物(R2)以及第一标签(t1)与第二反向引物(R2)之间的第一正向引物的5’端部分序列或全长序列(F1^),(e)混合第一和第二靶核酸片段、第一、第二、第三和第四核酸序列以及有效量的进行聚合酶链式反应(PCR)必需的试剂;以及(f)进行PCR。
F1、R1、F2、R2是基因特异性引物,其与基因组DNA的特定区域(靶DNA或扩增子)互补。这些引物的长度可由本领域技术人员选择。通常,这些基因特异性引物的长度是6-40、10-50或10-100个核苷酸。例如,这些基因特异性引物可以是15-30个核苷酸。
F1^是在R2引物的5’端处具有标签的F1引物序列的5’部分;F1^可以是F1的全长序列或部分序列。F1^的长度可取决于其GC含量,这影响其与互补碱基杂交时的解链点(melting point)。在一个实施方式中,F2^的部分序列比F2短1-20、1-10或1-5个核苷酸。在一个实施方式中,F2^的部分序列含有F2序列的10-50%、20-80%、30-70%、40-90%或50-90%。在另一个实施方式中,F2^的部分序列含有3-30或5-20或8-15个核苷酸。
标签t1、t2和t3是不结合靶DNA的通用标签序列。在一个实施方式中,标签t2和t3具有相同序列。在另一个实施方式中,t2和t3是不同的,即它们不是100%相同的。标签t2和t3均与标签t1不同。各标签均在基因特异性引物的5’端。在本发明中,这些标签序列的长度至少为3个核苷酸,且可以是长度为5-100、3-40或10-30个核苷酸。标签通常设计为使基因特异性无标签引物的解链温度增加至少5℃。标签序列可以是修饰的或未修饰的核酸。许多修饰的碱基(例如锁核酸或肽核酸)的退火温度高于其相应的天然碱基。当出于多种原因需要使用较短的标签序列时,这些修饰的碱基可用于代替天然碱基。
图1A和1B用于例示目的,不表明本发明仅限于这些附图。图1A显示使用不具有重叠区域的两条靶序列的典型PCR扩增。图1B显示F1和R2引物的PD形成和茎环结构对PD累积的抑制。
图1B显示本发明如何防止引物二聚体的指数扩增。在图1B中,正向引物F1和反向引物R2在其3’端具有互补区域。循环1后,形成PD链1和PD链2。循环2中,在左侧,PD链2形成茎环,其中t1和F1^分别退火至其互补对应物以形成茎,且剩余的核苷酸形成环。由于t1和F1^及其各自互补对应物的高局部浓度,即它们在同一PD链2上且彼此靠近,形成茎环比与单独的t1F1引物退火更为有利。因此,阻断了进一步的引物退火,且不会获得PD链2的进一步扩增产物。F1^的存在是重要的,其旨在完全阻断引物(t1_F1)退火至PD链2和随后的PD链2的扩增。在不存在F1^的情况下,引物t1_F1可优先于仅含t1的茎环并随后退火至PD链2。通过添加F1^,对于退火至PD链2,引物t1_F1不再优先于含有t1_F1^的茎环。
图1B的循环2中,在右侧,与PD链2类似,PD链1也形成茎环,其中t1和F1^分别退火至其互补对应物以形成茎,且剩余的核苷酸形成环。由于带标签的R2引物(t1_F1^_R2)的长度较长且因此解链温度较高,该引物可优先于茎中的t1_F1^进行退火,且可针对PD链1获得可能的线性扩增。图1B显示具有t1F1和t1_F1^_R2的引物设计的发明,PD将最多针对一条链进行线性扩增,且不会被指数扩增。
在本发明的步骤(f)中,可以一阶段(一种循环条件)或两阶段(两种不同的循环条件)的形式进行PCR。在两阶段PCR中,退火温度在第二循环条件中增加,其进一步减少引物二聚体形成。
在一阶段中,PCR包括以下步骤:(f1)激活DNA聚合酶并使(e)的混合物中的DNA变性,以及(f2)通过PCR的变性、退火和引物延伸步骤对(f1)的混合物循环多次以获得扩增产物。
在两阶段中,PCR包括以下步骤:(f-i)激活DNA聚合酶并使(e)的混合物中的DNA变性,(f-ii)通过PCR的变性、退火和引物延伸步骤对(f-i)的混合物循环至少两次,以及(f-iii)在高于步骤(f-ii)中退火温度的退火温度下通过PCR的变性、退火和引物延伸步骤对(f-i)的混合物进行循环以获得扩增产物。
两阶段PCR中,在步骤(f-ii)中,核酸和试剂的混合物通过变性、退火和引物延伸的PCR循环运行至少两次,如2-5次。在步骤(f-iii)中,(f)的混合物经历更多次变性、退火和引物延伸的PCR循环;此时是在高于步骤(f-ii)中退火温度的退火温度下进行的。例如,步骤(f-iii)中的退火温度比步骤(f-ii)中的退火温度高约4-35℃,或5-25℃,或6-20℃,或6-15℃。例如,退火和延伸的第一循环(步骤f-ii)的第一温度是58-62℃,例如60℃,且退火和延伸的第二循环(步骤f-iii)的第二温度是66-70℃,例如68℃。
两阶段PCR中,提高第二阶段(f-iii)中的退火温度以防止引物二聚体的反复起始。PCR的第一阶段(f-ii)后,两端的标签使得各扩增的靶序列产物延长且因此标签使得退火区域延长。因此,提高第二阶段中的退火温度降不会影响引物退火至特异性靶DNA。然而,提高第二阶段中的退火温度将减少引物二聚体的起始,其中互补区域保持相同长度。
以下实施例将进一步说明本发明。这些实施例仅旨在说明本发明而不应理解为限制本发明。
实施例
表1显示用于以下实施例的寡核苷酸序列。
表1
*小写字母表示标签序列;下划线表示R2中插入的部分F1序列;未标记的大写序列是基因特异性序列。
表1中的寡核苷酸1-4是不具有标签序列的针对BRCA1基因扩增子1和扩增子2的靶标特异性引物;扩增子1和扩增子2不具有重叠序列。寡核苷酸5-6是标签序列,其来自亿明达公司(Illumina)TSCA标签序列。寡核苷酸7-15是实施例1-3中使用的带标签的引物。
F1和R2在其3’端具有7个互补碱基且形成图2所示的杂合二聚体。
表2显示包括PD的扩增子大小,和人基因组19上的位置。
表2
*大小仅反映使用具有t1和t2标签且不具有F1^的引物的扩增子。
表3显示实施例1、2和3中引物组合的信息。
表3
*含有F1和R2的相互作用的引物以粗体显示于2重PCR引物混合物中
实施例1:1阶段PCR扩增
基因特异性PCR的典型的25μL PCR反应混合物包括:2μL人基因组DNA(普洛麦格公司(Promega)目录号G3041,使用低TE缓冲液(USB公司目录号75793)稀释至5ng/μL)、12.5μL的2x主混合物(Master Mix)(凯杰公司(Qiagen)目录号206413)、8.5μL无核酸酶水,和2μL的基因特异性引物混合物(各2.5μM,混合信息参见表3,寡核苷酸序列参见表1)。
1重和2重PCR反应均如下所述在热循环仪上进行:
1次循环 95℃15分钟酶激活和初始DNA变性
30次循环 95℃30秒 变性
60℃90秒 退火/延伸
1次循环 72℃5分钟 最终延伸
1次循环 8℃保持
在该实施例中,退火和延伸温度在循环期间保持恒定;因此其称作1阶段PCR扩增。
实施例2:2阶段PCR扩增
使用与实施例1类似的PCR反应混合物,但在热循环仪上使用2阶段PCR循环方案。前五次退火和延伸的循环在60℃下进行,这与实施例1中使用的温度相同。后续的25次退火和延伸的循环在升高的68℃下进行以抑制引物二聚体的起始。
该2阶段PCR方案如下所示:
1次循环 95℃15分钟 酶激活和初始DNA变性
5次循环 95℃30秒 变性
60℃90秒 退火/延伸
25次循环 95℃30秒 变性
68℃90秒 在升高的温度下进行退火/延伸
1次循环 72℃5分钟 最终延伸
1次循环 8℃保持
实施例3:琼脂糖凝胶电泳
在E-Base设备(生命科学技术公司(Life Technologies))上分析PCR产物。将2μL的各PCR产物与18μL无核酸酶水混合并随后直接加载至2%E-凝胶(E-gel)上。对稀释的PCR产物和50bp DNA梯状条带(英杰公司(Invitrogen)目录号10488-043)进行DNA凝胶电泳。在运行的末端,通过E-凝胶成像仪(E-gel Imager)(生命科学技术公司)捕获凝胶的数字图像。结果示于图3。
图3中,顶部小图显示1阶段PCR方案(实施例1)的结果,底部小图显示2阶段PCR方案(实施例2)的结果。泳道1和2是1重PCR,显示靶扩增子1和2的大小。剩余的反应都是2重PCR(泳道3-8)。当这两种扩增子多路复合在一起时,由于F1和R2的3’端的强相互作用,在1阶段和2阶段PCR条件下均形成F1+R2二聚体扩增子并且其在PCR反应物中占主导(如泳道3所示)。泳道4-8中PD中形成的茎结构除F1序列的5’端部分的3、6、9、12和15个核苷酸以外还含有t1序列(20nt)。与泳道3(无F1序列)相比,导入部分F1序列减少了泳道4-8中检测到的二聚体含量。当二聚体扩增被充分抑制时,靶扩增子成为可检测的(上方小图中的泳道6和下方小图中的泳道5)。当两个小图中的泳道7-8中实现二聚体扩增的近乎完全抑制时,两种靶扩增子产物在反应物中占主导。
目前以这类完整、清楚、简洁和准确的形式描述了本发明以及制备和使用的方式和方法,使得本领域技术人员对其进行制备和使用。应理解,前文描述了本发明的优选实施方式且可作出修改而不背离权利要求所涵盖的本发明的范围。为具体指出并明确要求本发明的主题,以下权利要求对说明书做出总结。
- 还没有人留言评论。精彩留言会获得点赞!