聚碳酸酯系树脂的发泡颗粒和发泡成形体的制作方法
本发明涉及聚碳酸酯系树脂的发泡颗粒和发泡成形体。更具体地,本发明涉及在发泡性、成形性和外观方面良好的聚碳酸酯系树脂的发泡成形体,和能够提供发泡成形体的聚碳酸酯系树脂的发泡颗粒。
背景技术:
除了发泡成形体的轻量性以外,由于其良好的加工性、形状保持性和相对大的强度,发泡成形体用于各种领域,所述领域包括食品托盘和汽车部件,并且还包括建筑材料、土木工程材料和照明器具。存在如下倾向:聚苯乙烯系树脂制的发泡成形体用于其中不特别要求耐热性的情况,并且例如聚丙烯和聚乙烯等烯烃系树脂制的发泡成形体用于其中要求缓冲特性、恢复性和柔软性等的情况。
一般具有比聚苯乙烯系树脂和烯烃系树脂更高的耐热性的树脂的实例包括聚碳酸酯系树脂。聚碳酸酯系树脂为能够用于日本以外的国家和例如干旱地区和热带地区等在极端气候条件下的区域的树脂材料。聚碳酸酯系树脂不仅耐热性优异,而且在耐水性、电气特性、机械强度、耐老化性和耐化学品性方面也是优异的。由于这些特性,聚碳酸酯系树脂用作建筑物的内饰材料,并且近年来,期待通过利用其优异的特性扩展至例如汽车部件、包装材料和各种容器等这样的用途。
聚碳酸酯系树脂的发泡体的公知的制法包括专利文献1(日本专利特开平9-076332号公报)和专利文献2(日本专利特开平11-236736号公报)中记载的挤出发泡法。然而,通过该方法获得的发泡体为板(board)状,由其仅可以获得简单的建筑材料。因此,难以通过挤出发泡法提供例如汽车部件等具有复杂的形状的发泡体。
使得复杂的形状成为可能的已知的方法包括模具内发泡成形法,其中使发泡颗粒在模具内发泡和熔合(fusionbonded)。在该方法中,准备具有对应于期望的形状的空间的模具,所述空间内填充有发泡颗粒,并且通过加热使发泡颗粒发泡和熔合,由此使得可以制造具有复杂的形状的发泡成形体。在专利文献3(日本专利特开平6-100724号公报)、专利文献4(日本专利特开平11-287277号公报)和专利文献5(国际公开wo2011/019057号)中提出用于从由聚碳酸酯系树脂制成的发泡颗粒通过模具内发泡成形法提供发泡成形体的方法。
引文列表
专利文献
专利文献1:日本专利特开平9-076332号公报
专利文献2:日本专利特开平11-236736号公报
专利文献3:日本专利特开平6-100724号公报
专利文献4:日本专利特开平11-287277号公报
专利文献5:国际公开wo2011/019057号
技术实现要素:
发明要解决的问题
然而,在专利文献3~5中存在如下问题:包括在高发泡化方面的困难和不利的成形性,并且还包括发泡成形体的劣化的外观。
此外,在专利文献3~5中,在提供发泡成形体时,通过用蒸气加热使发泡颗粒熔合,因此难以提供具有彼此充分地熔合的发泡颗粒的发泡成形体。虽然可能存在使用粘接剂用于粘接发泡颗粒的方法,但是可以容易地推测,其使用变为使轻量化和聚碳酸酯系树脂固有的热特性劣化的因素。
用于解决问题的方案
鉴于上述问题,作为本发明的发明人对所使用的聚碳酸酯系树脂的研究的结果,惊奇地发现,通过使用如下聚碳酸酯系树脂,可以提供在发泡性、成形性和外观方面良好的发泡成形体,和能够提供发泡成形体的聚碳酸酯系树脂的发泡颗粒:所述聚碳酸酯系树脂在通过借助反应热解gc/ms法(reactivepyrolysisgc/msmethod)的测量获得的gc/ms图中的特定的位置处具有(a)源自145~230的分子量的峰和源自320~350的分子量的峰、(b)源自210~230的分子量的峰、或(c)源自290~320的分子量的峰,并且由此完成了本发明。
本发明提供聚碳酸酯系树脂的发泡颗粒,其包含含有源自双酚a的组分的聚碳酸酯系树脂作为基材树脂,
在以保留时间作为横轴的gc/ms图中,所述发泡颗粒满足以下条件(a)~(c)中的任一项,所述gc/ms图通过在氦气用作载气且载气流量为34ml/分的条件下借助利用使用三甲基氢氧化铵作为反应试剂使包含在所述聚碳酸酯系树脂中的酯键通过水解而甲基醚化的反应的反应热解gc/ms法的测量来获得:
(a)显示源自145~230的分子量的峰和源自320~350的分子量的峰,
以显示所述源自双酚a的组分的最大峰的保留时间为基准,在-15分钟以内的范围内的保留时间,确认到所述源自145~230的分子量的峰,并且
以显示所述源自双酚a的组分的最大峰的保留时间为基准,在+10分钟以内的范围内的保留时间,确认到所述源自320~350的分子量的峰;
(b)显示源自210~230的分子量的峰,并且
以显示所述源自双酚a的组分的最大峰的保留时间为基准,在5分钟以内的范围内的保留时间,确认到所述源自210~230的分子量的峰;并且
(c)显示源自290~320的分子量的峰,并且
以显示所述源自双酚a的组分的最大峰的保留时间为基准,在5分钟以内的范围内的保留时间,确认到所述源自290~320的分子量的峰。
本发明还提供聚碳酸酯系树脂的发泡成形体,其包含多个发泡颗粒,
所述发泡颗粒包含含有源自双酚a的组分的聚碳酸酯系树脂作为基材树脂,
在以保留时间作为横轴的gc/ms图中,所述发泡成形体满足以下条件(a)~(c)中的任一项,所述gc/ms图通过在氦气用作载气且载气流量为34ml/分的条件下借助利用使用三甲基氢氧化铵作为反应试剂使包含在所述聚碳酸酯系树脂中的酯键通过水解而甲基醚化的反应的反应热解gc/ms法的测量来获得:
(a)显示源自145~230的分子量的峰和源自320~350的分子量的峰,
以显示所述源自双酚a的组分的最大峰的保留时间为基准,在-15分钟以内的范围内的保留时间,确认到所述源自145~230的分子量的峰,并且
以显示所述源自双酚a的组分的最大峰的保留时间为基准,在+10分钟以内的范围内的保留时间,确认到所述源自320~350的分子量的峰;
(b)显示源自210~230的分子量的峰,并且
以显示所述源自双酚a的组分的最大峰的保留时间为基准,在5分钟以内的范围内的保留时间,确认到所述源自210~230的分子量的峰;并且
(c)显示源自290~320的分子量的峰,并且
以显示所述源自双酚a的组分的最大峰的保留时间为基准,在5分钟以内的范围内的保留时间,确认到所述源自290~320的分子量的峰。
发明的效果
根据本发明,可以提供在发泡性、成形性和外观方面良好的聚碳酸酯系树脂的发泡成形体,和能够提供发泡成形体的聚碳酸酯系树脂的发泡颗粒。
根据任意以下情况,可以提供在发泡性、成形性和外观方面进一步良好的聚碳酸酯系树脂的发泡成形体,和能够提供发泡成形体的聚碳酸酯系树脂的发泡颗粒。
(1)源自145~230的分子量的峰源自构成聚碳酸酯系树脂的末端部分,并且源自320~350的分子量的峰源自构成聚碳酸酯系树脂的支化结构部分。
(2)显示源自双酚a的组分的最大峰与源自145~230的分子量的峰的面积比为1/0.02~1/0.07,并且显示源自双酚a的组分的最大峰与源自320~350的分子量的峰的面积比为1/0.005~1/0.05。
(3)源自210~230的分子量的峰源自构成聚碳酸酯系树脂的末端部分。
(4)显示源自双酚a的组分的最大峰与源自210~230的分子量的峰的面积比为1/0.01~1/0.07。
(5)显示源自双酚a的组分的最大峰与源自290~320的分子量的峰的面积比为1/0.005~1/0.04。
(6)发泡颗粒的体积密度为0.08g/cm3以下。
(7)发泡颗粒的气泡密度x为1.0×108~1.0×1012个/cm3[其中气泡密度x通过下式来计算:
气泡密度x=(ρ/d-1)/{(4/3)·π·(c/10/2)3}
式中,c表示平均气泡直径(mm);ρ表示聚碳酸酯系树脂的密度(kg/m3);并且d表示发泡颗粒的表观密度(kg/m3)]。
(8)平均气泡直径为0.0030~0.2000mm,聚碳酸酯系树脂的密度为1.0×103~1.4×103kg/m3,并且发泡颗粒的表观密度为12~600kg/m3。
(9)发泡成形体的密度为0.08g/cm3以下。
此外,在以下情况下,即使聚碳酸酯系树脂在如上所述的特定的位置处不具有显示特定的分子量的峰,通过将发泡颗粒和发泡成形体的气泡密度x限定至特定的范围,也可以改善从发泡颗粒获得的发泡成形体的外观,并且可以提高发泡颗粒之间的熔合性。此外,由于发泡颗粒的特定的气泡密度x,因此可以在气泡膜的厚度方面控制发泡颗粒,由此提供改善成形时的二次发泡性的效果。
包含聚碳酸酯系树脂作为基材树脂的发泡颗粒,
所述发泡颗粒的气泡密度x为1.0×108~1.0×1012个/cm3[其中气泡密度x通过下式来计算:
气泡密度x=(ρ/d-1)/{(4/3)·π·(c/10/2)3}
式中,c表示平均气泡直径(mm);ρ表示聚碳酸酯系树脂的密度(kg/m3);并且d表示发泡颗粒的表观密度(kg/m3)]。
包含聚碳酸酯系树脂作为基材树脂的发泡成形体,
所述发泡成形体的气泡密度x为1.0×108~1.0×1012个/cm3[其中气泡密度x通过下式来计算:
气泡密度x=(ρ/d-1)/{(4/3)·π·(c/10/2)3}
式中,c表示平均气泡直径(mm);ρ表示聚碳酸酯系树脂的密度(kg/m3);并且d表示发泡成形体的表观密度(kg/m3)]。
此外,在以下情况下,即使聚碳酸酯系树脂在如上所述的特定的位置处不具有显示特定的分子量的峰,通过使用具有特定的结构(包括酯键数和分子量)和特定的沸点的增塑剂,也可以提供熔合性优异的发泡成形体。
一种聚碳酸酯系树脂的发泡成形体,其包含含有聚碳酸酯系树脂作为基材树脂的多个发泡颗粒,所述发泡成形体包含具有2个以上的酯键、分子量为200~600且沸点为250~500℃的增塑剂。
附图说明
[图1]为示出用反应热解gc/ms测量实施例1a和实施例1b的发泡颗粒的结果的图。
[图2]为示出用反应热解gc/ms测量实施例2a的发泡颗粒的结果的图。
[图3]为示出用反应热解gc/ms测量实施例3a的发泡颗粒的结果的图。
[图4]为示出用反应热解gc/ms测量实施例2b的发泡颗粒的结果的图。
[图5]为示出用反应热解gc/ms测量实施例1c的发泡颗粒的结果的图。
[图6]为实施例1d的发泡颗粒的切断面的照片。
[图7]为实施例2d的发泡颗粒的切断面的照片。
[图8]为实施例3d的发泡颗粒的切断面的照片。
[图9]为实施例1d的发泡成形体的切断面的照片。
具体实施方式
(聚碳酸酯系树脂的发泡颗粒)
聚碳酸酯系树脂的发泡颗粒(下文中可以简称为发泡颗粒)包含满足条件(a)~(c)中的任一项的含有源自双酚a的组分的聚碳酸酯系树脂(a)~(c)中的任一种作为基材树脂。
·聚碳酸酯系树脂(a)
[源自145~230的分子量的峰]
发泡颗粒在gc/ms图中显示源自145~230的分子量的峰。以显示源自双酚a的组分的最大峰(下文中可以称为最大峰)的保留时间为基准,在-15分钟以内的范围内的保留时间确认到所述峰。通过在氦气用作载气且载气流量为34ml/分的条件下,借助利用使用三甲基氢氧化铵作为反应试剂使包含在聚碳酸酯系树脂中的酯键通过水解而甲基醚化的反应的反应热解gc/ms法的测量获得gc/ms图。
本发明人惊奇地发现,通过以最大峰的保留时间为基准,在-15分钟以内的范围内的保留时间确认到源自145~230的分子量的峰,可以提供能够提供在成形性和外观方面良好的聚碳酸酯系树脂的发泡成形体(下文中可以简称为发泡成形体)的发泡颗粒。本发明人推测,提供在成形性和外观方面良好的发泡成形体的机理如下。
具体地,源自145~230的分子量的峰显示聚碳酸酯系树脂的特有的结构。本发明人发现,具有对应于所述峰的特有的结构的聚碳酸酯系树脂在实施例中提供在成形性和外观方面良好的发泡成形体。所述峰可归因于聚碳酸酯系树脂的末端结构。末端结构使构成聚碳酸酯系树脂的高分子链缠结,从而提供具有由多个高分子链形成的空隙的聚碳酸酯系树脂。空隙有助于发泡剂的保持性的提高从而使高分子链延伸,并且有助于发泡剂的耗散性(dissipation),由此提供能够提供在发泡时成形性和外观方面良好的发泡成形体的发泡颗粒。
本发明人推测,源自145~230的分子量的峰为构成聚碳酸酯系树脂的末端部分。具体地,用于实施例的由sabic制造的lexan153具有以最大峰的保留时间为基准在-15分钟以内的范围内的保留时间确认到的源自210~230的分子量的峰。本发明人推测,所述峰可归因于2-(1-甲基-1-苯基乙基)苯酚。用于实施例的由teijinltd.制造的panlitex0730和由bayerag制造的makrolonwb1439各自具有以最大峰的保留时间为基准在-15分钟以内的范围内的保留时间确认到的源自145~165的分子量的峰。本发明人推测,所述峰可归因于1-(1,1-二甲基乙基)-4-甲氧基苯。
在源自所得gc/ms图的质谱中,显示源自双酚a的组分的最大峰与源自145~230的分子量的峰的面积比优选为1/0.02~1/0.07。峰的面积比可归因于末端数量和主链骨架的重复单元的数量,并且为影响聚合物的运动的容易性和硬度等的值。在其中源自145~230的分子量的峰的峰面积比小于0.02的情况下,由于聚合物链的大的长度而导致在发泡时聚合物链的移动会减慢,由此在一些情况下不能良好地进行发泡。在其中源自145~230的分子量的峰的峰面积比大于0.07的情况下,由于聚合物链的小的长度而导致气泡不能耐受发泡剂的膨胀,因此气泡会在发泡之后立即收缩,由此在一些情况下不能良好地进行发泡。源自145~230的分子量的峰的面积比的值可以为0.02、0.03、0.04、0.05、0.06、0.065或0.07。源自145~230的分子量的峰的面积比的下限更优选为0.022,并且进一步优选0.025。源自145~230的分子量的峰的面积比的上限更优选为0.065,并且进一步优选0.060。
更具体地,显示源自双酚a的组分的最大峰与源自210~230的分子量的峰的面积比优选为1/0.03~1/0.06。源自210~230的分子量的峰的面积比的值可以为0.03、0.04、0.05或0.06。显示源自双酚a的组分的最大峰与源自145~165的分子量的峰的面积比优选为1/0.015~1/0.05。源自145~165的分子量的峰的面积比的值可以为0.015、0.02、0.03、0.04或0.05。
[源自320~350的分子量的峰]
发泡颗粒在gc/ms图中显示源自320~350的分子量的峰。以显示源自双酚a的组分的最大峰(下文中可以称为最大峰)的保留时间为基准,在+10分钟以内的范围内的保留时间,确认到所述峰。
本发明人惊奇地发现,与对于源自145~230的分子量的峰记载的前述情况类似,当以最大峰的保留时间为基准在+10分钟以内的范围内的保留时间确认到源自320~350的分子量的峰时,可以提供能够提供在成形性和外观方面良好的发泡成形体的发泡颗粒。本发明人推测,提供在成形性和外观方面良好的发泡成形体的机理如下。
具体地,源自320~350的分子量的峰显示聚碳酸酯系树脂的特有的结构。本发明人发现,具有对应于所述峰的特有的结构的聚碳酸酯系树脂在实施例中提供在成形性和外观方面良好的发泡成形体。所述峰可归因于聚碳酸酯系树脂的支化结构。推测与末端结构类似,支化结构不仅在发泡期间有助于气体耗散性,而且还特别地在成形期间促进分子间的缠结,由此,显著地改善颗粒间的粘接性。
本发明人推测,源自320~350的分子量的峰为构成聚碳酸酯系树脂的支化结构部分,并且,特别地,可归因于2-三(对羟基苯基)乙酸酯。
在源自所得gc/ms图的质谱中,显示源自双酚a的组分的最大峰与源自320~350的分子量的峰的面积比优选为1/0.005~1/0.05。峰的面积比为显示主链骨架的支化程度的值,并且为影响聚合物的运动的容易性、气体耗散性和成形时的分子的缠结的值。在其中源自320~350的分子量的峰的峰面积比小于0.005的情况下,由于小的支化程度而导致缠结会是小的,由此在一些情况下不能良好地进行发泡并且不能有利地提供发泡成形体。在其中源自320~350的分子量的峰的峰面积比大于0.05的情况下,由于大量的支化结构,因此在使聚合物软化时,聚合物分子会难以彼此分离并且难以移动,由此在一些情况下不能良好地进行发泡。源自320~350的分子量的峰的面积比的值可以为0.005、0.008、0.01、0.02、0.03、0.04、0.045或0.05。源自320~350的分子量的峰的面积比的下限更优选为0.008,并且进一步优选0.01。源自320~350的分子量的峰的面积比的上限更优选为0.045,并且进一步优选0.04。
·聚碳酸酯系树脂(b)
[源自210~230的分子量的峰]
发泡颗粒在gc/ms图中显示源自210~230的分子量的峰。可以通过以与聚碳酸酯系树脂(a)中相同的方式的测量来获得所述峰。
本发明人惊奇地发现,当以最大峰的保留时间为基准在5分钟以内的范围内的保留时间确认到源自210~230的分子量的峰时,可以提供能够提供在成形性和外观方面良好的发泡成形体的发泡颗粒。本发明人推测,提供在成形性和外观方面良好的发泡成形体的机理如下。
具体地,源自210~230的分子量的峰显示聚碳酸酯系树脂的特有的结构。本发明人发现,具有对应于所述峰的特有的结构的聚碳酸酯系树脂在实施例中提供在成形性和外观方面良好的发泡成形体。所述峰可归因于聚碳酸酯系树脂的末端结构。末端结构使构成聚碳酸酯系树脂的高分子链缠结,从而提供具有由多个高分子链形成的空隙的聚碳酸酯系树脂。空隙有助于发泡剂的保持性的提高从而使高分子链延伸,并且有助于发泡剂的耗散性,由此提供能够提供在发泡时成形性和外观方面良好的发泡成形体的发泡颗粒。
本发明人推测,源自210~230的分子量的峰为构成聚碳酸酯系树脂的末端部分,并且,特别地,推测所述峰可归因于2-(1-甲基-1-苯基乙基)苯酚或4-叔丁基-2-苯基苯酚。
在源自所得gc/ms图的质谱中,显示源自双酚a的组分的最大峰与源自210~230的分子量的峰的面积比优选为1/0.01~1/0.07。峰的面积比可归因于末端数量和主链骨架的重复单元的数量,并且为影响聚合物的运动的容易性和硬度等的值。在其中源自210~230的分子量的峰的峰面积比小于0.01的情况下,由于聚合物链的大的长度而导致在发泡时聚合物链的运动会减慢,由此在一些情况下不能良好地进行发泡。在其中源自210~230的分子量的峰的峰面积比大于0.07的情况下,由于聚合物链的小的长度而导致气泡不能耐受发泡剂的膨胀,因此气泡会在发泡之后立即收缩,由此在一些情况下不能良好地进行发泡。源自210~230的分子量的峰的面积比的值可以为0.01、0.015、0.02、0.03、0.04、0.05、0.06或0.07。源自210~230的分子量的峰的面积比的下限更优选为0.015,进一步优选0.02,并且特别优选0.03。源自210~230的分子量的峰的面积比的上限更优选为0.06。
·聚碳酸酯系树脂(c)
[源自290~320的分子量的峰]
发泡颗粒在gc/ms图中显示源自290~320的分子量的峰。可以通过以与聚碳酸酯系树脂(a)中相同的方式的测量来获得所述峰。
本发明人惊奇地发现,当以最大峰的保留时间为基准在5分钟以内的范围内的保留时间确认到源自290~320的分子量的峰时,可以提供在成形性和外观方面良好的发泡成形体。本发明人推测,提供在成形性和外观方面良好的发泡成形体的机理如下。
具体地,源自290~320的分子量的峰显示聚碳酸酯系树脂的特有的结构。本发明人发现,具有对应于所述峰的特有的结构的聚碳酸酯系树脂在实施例中提供在成形性和外观方面良好的发泡成形体。推测所述峰可归因于聚碳酸酯系树脂的支化结构。支化结构使构成聚碳酸酯系树脂的高分子链缠结,从而提供具有由多个高分子链形成的空隙的聚碳酸酯系树脂。空隙有助于发泡剂的保持性的提高从而使高分子链延伸,并且有助于发泡剂的耗散性。
在源自所得gc/ms图的质谱中,显示源自双酚a的组分的最大峰与源自290~320的分子量的峰的面积比优选为1/0.005~1/0.04。峰的面积比可归因于分支数量和主链骨架的重复单元的数量,并且为影响聚合物的运动的容易性和硬度等的值。在其中源自290~320的分子量的峰的峰面积比小于0.005的情况下,由于少量的支化结构而导致在发泡时聚合物的缠结会是少的,由此在一些情况下不能良好地提供发泡成形体。在其中源自290~320的分子量的峰的峰面积比大于0.04的情况下,由于大量的支化结构而导致聚合物在发泡时会难以移动,这会导致气泡的破裂和收缩等,由此在一些情况下不能良好地进行发泡并且不能提供成形体。源自290~320的分子量的峰的面积比的值可以为0.005、0.01、0.015、0.02、0.03或0.04。
峰面积比的上限优选为0.03,更优选0.02,并且进一步优选0.015。
[聚碳酸酯系树脂]
聚碳酸酯系树脂优选具有碳酸与二元醇或二元酚的聚酯结构。从进一步提高耐热性的观点,聚碳酸酯系树脂优选具有芳香族骨架。聚碳酸酯系树脂的具体实例包括源自双酚的聚碳酸酯树脂,如2,2-双(4-氧苯基)丙烷、2,2-双(4-氧苯基)丁烷、1,1-双(4-氧苯基)环己烷、1,1-双(4-氧苯基)丁烷、1,1-双(4-氧苯基)异丁烷、和1,1-双(4-氧苯基)乙烷。
其中,优选(i)具有源自2-(1-甲基-1-苯基乙基)苯酚或1-(1,1-二甲基乙基)-4-甲氧基苯的组分作为末端部分和源自2-三(对羟基苯基)乙酸酯的组分作为支化结构部分的树脂、和(ii)具有源自2-(1-甲基-1-苯基乙基)苯酚或4-叔丁基-2-苯基苯酚的组分作为末端部分的树脂,所述(i)和(ii)各自具有源自2,2-双(4-氧苯基)丙烷(俗名:双酚a)的组分作为基本骨架。
聚碳酸酯系树脂可以包含除了聚碳酸酯树脂以外的其它树脂。其它树脂的实例包括丙烯酸系树脂、饱和聚酯树脂、abs树脂、聚苯乙烯树脂、和聚苯醚树脂。可以包含不具有任意前述末端结构和支化结构的聚碳酸酯系树脂。聚碳酸酯系树脂优选以50重量%以上的量包含具有前述末端结构和支化结构的聚碳酸酯系树脂。
聚碳酸酯系树脂的mfr优选为1~20g/10min。在该范围内的树脂适合于发泡,并且可以进一步高发泡化。mfr更优选在2~15g/10min的范围内。
发泡颗粒可以包含聚碳酸酯系树脂作为基材树脂并且可以具有特定的气泡密度x。从实施例与比较例之间的比较可以明显看出,气泡密度x为相对高的值。本发明人发现,可以通过增加气泡密度x来提高发泡成形体的外观和熔合性。即使聚碳酸酯系树脂在如上所述的特定的位置处不具有显示特定的分子量的峰,也可以显示提高效果。可以通过具有所述峰来进一步增强提高效果。
气泡密度x可以为1.0×108~1.0×1012个/cm3。在其中气泡密度x小于1.0×108个/cm3的情况下,气泡膜会变厚,从而在一些情况下使成形前的二次发泡性降低。在其中气泡密度x大于1.0×1012个/cm3的情况下,气泡膜会变薄,从而促进在发泡时气泡膜的破裂,在一些情况下提供连续的气泡。气泡密度x的值可以为1.0×108个/cm3、1.2×108个/cm3、1.5×108个/cm3、1.0×109个/cm3、1.0×1010个/cm3、1.0×1011个/cm3、5.0×1011个/cm3或1.0×1012个/cm3。气泡密度x优选为1.2×108~5.0×1011个/cm3,并且更优选为1.5×108~1.0×1011个/cm3。
气泡密度x可以通过下式来计算:
气泡密度x=(ρ/d-1)/{(4/3)·π·(c/10/2)3}
式中,c表示平均气泡直径(mm);ρ表示聚碳酸酯系树脂的密度(kg/m3);并且d表示发泡颗粒的表观密度(kg/m3)。
平均气泡直径c优选在0.0030~0.2000mm的范围内。平均气泡直径c的值可以为0.0030mm、0.0034mm、0.0057mm、0.0100mm、0.0500mm、0.0850mm、0.0910mm、0.1000mm、0.1500mm、0.1800mm或0.2000mm。平均气泡直径c更优选为0.0034~0.0910mm,并且平均气泡直径c进一步优选为0.0057~0.0850mm。
聚碳酸酯系树脂的密度ρ优选在1.0×103~1.4×103kg/m3的范围内。在其中密度ρ小于1.0×103kg/m3的情况下,耐热温度在一些情况下会降低。在其中密度ρ大于1.4×103kg/m3的情况下,耐热温度在一些情况下上升从而使发泡成形困难。密度ρ的值可以为1.0×103kg/m3、1.10×103kg/m3、1.15×103kg/m3、1.20×103kg/m3、1.30×103kg/m3、1.35×103kg/m3或1.4×103kg/m3。密度ρ更优选为1.10×103~1.35×103kg/m3,并且密度ρ进一步优选为1.15×103~1.30×103kg/m3。
发泡颗粒的表观密度d优选在12~600kg/m3的范围内。在其中表观密度d小于12kg/m3的情况下,气泡膜会变薄,气泡膜在二次发泡时会破裂,连续气泡的比例会增加,并且在一些情况下会发生由于气泡的弯曲而导致的发泡颗粒的收缩等。在其中表观密度d大于600kg/m3的情况下,气泡膜会变厚,并且二次发泡性在一些情况下会劣化。表观密度d的值可以为12kg/m3、24kg/m3、30kg/m3、50kg/m3、100kg/m3、120kg/m3、240kg/m3或600kg/m3。表观密度d更优选为24~240kg/m3,并且表观密度d进一步优选为30~120kg/m3。
[发泡颗粒的形状]
对发泡颗粒的形状不特别限定。其形状的实例包括球状和圆柱状。其中,其形状优选尽可能接近球状。换言之,发泡颗粒的短径与长径的比优选尽可能接近1。
发泡颗粒可以具有各种体积密度。体积密度优选为0.4g/cm3以下。体积密度的值可以为0.01g/cm3、0.012g/cm3、0.04g/cm3、0.06g/cm3、0.12g/cm3、0.3g/cm3或0.4g/cm3。体积密度更优选为0.010~0.12g/cm3,进一步优选0.012~0.12g/cm3,并且特别优选0.01~0.08g/cm3。体积密度可以为0.04g/cm3以上,并且可以为0.06~0.3g/cm3。
发泡颗粒的平均粒径优选为1~20mm。
[发泡颗粒的制造方法]
可以通过用发泡剂浸渍树脂颗粒从而提供发泡性颗粒和使发泡性颗粒发泡来获得发泡颗粒。
(1)发泡性颗粒的制造
可以通过用发泡剂浸渍聚碳酸酯系树脂制的树脂颗粒来获得发泡性颗粒。
可以通过已知的方法获得树脂颗粒。其实例包括如下方法:其中将聚碳酸酯系树脂与根据需要的添加剂一起在挤出机中熔融混炼,由其挤出成线料(strand),并且将所得线料在空气中切割、在水中切割、或者在加热下切割,从而将树脂造粒。使用的树脂颗粒可以为商购可得的树脂颗粒。树脂颗粒可以根据需要包含除了树脂以外的其它添加剂。添加剂的实例包括增塑剂、阻燃剂、阻燃助剂、结合抑制剂、抗静电剂、铺展剂、气泡调节剂、填料、着色剂、耐候剂、防老剂、润滑剂、防雾剂和香料。
结合抑制剂(聚结抑制剂)在发泡步骤中具有抑制发泡颗粒的聚结的作用。本文中提及的聚结意指使多个发泡颗粒结合和一体化。结合抑制剂的具体实例包括滑石、碳酸钙和氢氧化铝。
抗静电剂的实例包括聚氧乙烯烷基酚醚和硬脂酸单甘油酯。
铺展剂的实例包括聚丁烯、聚乙二醇和硅油。
用来浸渍树脂颗粒的发泡剂可以为已知的挥发性发泡剂或已知的无机发泡剂。挥发性发泡剂的实例包括例如丙烷、丁烷和戊烷等脂肪族烃、芳香族烃、脂环式烃、和脂肪族醇。无机发泡剂的实例包括二氧化碳气体、氮气和空气(空气(air))。发泡剂可以以其两种以上的组合使用。在发泡剂中,无机发泡剂是优选的,并且二氧化碳气体是更优选的。
发泡剂的含量(浸渍量)相对于100重量份的聚碳酸酯系树脂优选为3~15重量份。在其中发泡剂的含量小于3重量份的情况下,发泡力会降低,由此在一些情况下不能良好地进行发泡。在其中发泡剂的含量超过15重量份的情况下,增塑化效果会增加,并且在发泡时倾向于发生收缩,由此在一些情况下使生产性劣化并且抑制期望的发泡倍率的稳定的实现。发泡剂的含量更优选为4~12重量份。
浸渍方法的实例包括将在搅拌下分散在水系中的树脂颗粒通过将发泡剂压入其中来浸渍的湿式浸渍法、和将树脂颗粒投入可密闭的容器中并且通过将发泡剂压入其中来浸渍的实质上不使用水的干式浸渍法(气相浸渍法)。能够在不使用水的情况下进行浸渍的干式浸渍法是特别优选的。对用发泡剂浸渍树脂颗粒时的浸渍压力、浸渍时间和浸渍温度不特别限定。
从有效浸渍从而提供更良好的发泡颗粒和发泡成形体的观点,浸渍压力优选为1~4.5mpa。
浸渍时间优选为0.5~200小时。在其中浸渍时间小于0.5小时的情况下,发泡剂向树脂颗粒的浸渍量会降低,由此在一些情况下不能提供充分的发泡力。在其中浸渍时间长于200小时的情况下,生产性在一些情况下会劣化。浸渍时间更优选为1~100小时。
浸渍温度优选为0~60℃。在其中浸渍温度低于0℃的情况下,由于在一些情况下在期望的时间内不能确保充分的浸渍量,因此不会获得充分的发泡力(一次发泡力)。在其中浸渍温度高于60℃的情况下,生产性在一些情况下会劣化。浸渍温度更优选为5~50℃。
(2)发泡颗粒的制造
通过使发泡性颗粒发泡来提供发泡颗粒的方法优选为通过用蒸气(水蒸气)等加热来使发泡性颗粒发泡的方法。
在发泡时的发泡机中优选使用密闭耐压的发泡容器。蒸气的压力优选为0.1~0.8mpa(表压),更优选0.2~0.5mpa,并且进一步优选0.25~0.45mpa。只要发泡时间为对于提供期望的发泡倍率是必要的时间就足够了。发泡时间优选为5~180秒。当发泡时间超过180秒时,发泡颗粒在一些情况下会开始收缩,并且在一些情况下不能从发泡颗粒获得具有良好的物性的发泡成形体。
在发泡颗粒的制造过程中,可以通过控制浸渍条件(例如,浸渍压力、浸渍时间和浸渍温度)和一次发泡条件(发泡压力和发泡时间)来使气泡密度x增大或减小。
(发泡成形体)
发泡成形体包含多个发泡颗粒,并且发泡颗粒包含满足条件(a)~(c)中的任一项的聚碳酸酯系树脂(a)~(c)中的任一种作为基材树脂。通常可以从前述发泡颗粒制造发泡成形体。
·聚碳酸酯系树脂(a)
[源自145~230的分子量和320~350的分子量的峰]
发泡成形体在以保留时间作为横轴的gc/ms图中显示源自145~230的分子量的峰。以最大峰的保留时间为基准,在-15分钟以内的范围内的保留时间,确认到所述峰。此外,发泡成形体在以保留时间作为横轴的gc/ms图中显示源自320~350的分子量的峰。以最大峰的保留时间为基准,在+10分钟以内的范围内的保留时间,确认到所述峰。确认方法与发泡颗粒的确认方法相同。
由于与发泡颗粒中相同的原因,因此在源自所得gc/ms图的质谱中,显示源自双酚a的组分的最大峰与源自145~230的分子量的峰的面积比优选为1/0.02~1/0.07。源自145~230的分子量的峰的面积比的值可以为0.02、0.03、0.04、0.05、0.06、0.065或0.07。源自145~230的分子量的峰的面积比的下限更优选为0.022,并且进一步优选0.025。源自145~230的分子量的峰的面积比的上限更优选为0.065。此外,显示源自双酚a的组分的最大峰与源自320~350的分子量的峰的面积比优选为1/0.005~1/0.05。源自320~350的分子量的峰的面积比的值可以为0.005、0.008、0.01、0.02、0.03、0.04、0.045或0.05。源自320~350的分子量的峰的面积比的下限更优选为0.008,并且进一步优选0.01。源自320~350的分子量的峰的面积比的上限更优选为0.045,并且进一步优选0.04。
·聚碳酸酯系树脂(b)
[源自210~230的分子量的峰]
发泡成形体在以保留时间作为横轴的gc/ms图中显示源自210~230的分子量的峰。以最大峰的保留时间为基准,在5分钟以内的范围内的保留时间,确认到所述峰。确认方法与发泡颗粒的确认方法相同。
由于与发泡颗粒中相同的原因,因此在源自所得gc/ms图的质谱中,显示源自双酚a的组分的最大峰与源自210~230的分子量的峰的面积比优选为1/0.01~1/0.07。源自210~230的分子量的峰的面积比的值可以为0.01、0.015、0.02、0.03、0.04、0.05、0.06或0.07。源自210~230的分子量的峰的面积比的下限更优选为0.015,进一步优选0.02,并且特别优选0.03。源自210~230的分子量的峰的面积比的上限更优选为0.06。
·聚碳酸酯系树脂(c)
[源自290~320的分子量的峰]
发泡成形体在以保留时间作为横轴的gc/ms图中显示源自290~320的分子量的峰。以最大峰的保留时间为基准,在5分钟以内的范围内的保留时间,确认到所述峰。确认方法与发泡颗粒的确认方法相同。
由于与发泡颗粒中相同的原因,因此在源自所得gc/ms图的质谱中,显示源自双酚a的组分的最大峰与源自290~320的分子量的峰的面积比优选为1/0.005~1/0.04。源自290~320的分子量的峰的面积比的值可以为0.005、0.01、0.015、0.02、0.03或0.04。源自290~320的分子量的峰的面积比的上限优选为0.03,更优选0.02,并且进一步优选0.015。
发泡成形体可以具有以下特定的气泡密度x。
由构成发泡成形体的发泡颗粒计算气泡密度x。与发泡颗粒类似,可以由下式计算气泡密度x:
气泡密度x=(ρ/d-1)/{(4/3)·π·(c/10/2)3}
其中d表示发泡成形体的表观密度(kg/m3)。
气泡密度x可以为1.0×108~1.0×1012个/cm3。气泡密度x在特定的范围内的原因与发泡颗粒的原因相同。气泡密度x的值可以为1.0×108个/cm3、1.2×108个/cm3、1.5×108个/cm3、1.0×109个/cm3、1.0×1010个/cm3、1.0×1011个/cm3、5.0×1011个/cm3或1.0×1012个/cm3。气泡密度x的优选的范围和更优选的范围分别与发泡颗粒的各范围相同。
平均气泡直径c和聚碳酸酯系树脂的密度ρ的优选的范围及其原因、更优选的范围、和进一步优选的范围分别与发泡颗粒的那些相同。平均气泡直径c的值可以为0.0030mm、0.0034mm、0.0057mm、0.0100mm、0.0500mm、0.0850mm、0.0910mm、0.1000mm、0.1500mm、0.1800mm或0.2000mm。
发泡成形体的密度d优选在12~600kg/m3的范围内。在其中表观密度d小于12kg/m3的情况下,气泡膜会变薄,气泡膜在二次发泡时会破裂,并且连续气泡的比例会增加,这在一些情况下会导致成形体的强度劣化。在其中表观密度d大于600kg/m3的情况下,气泡膜会变厚,并且二次发泡性和成形时的发泡颗粒的熔合性在一些情况下会劣化。表观密度d的值可以为12kg/m3、24kg/m3、30kg/m3、50kg/m3、100kg/m3、120kg/m3、240kg/m3或600kg/m3。表观密度d更优选为24~240kg/m3,并且表观密度d进一步优选为30~120kg/m3。
[增塑剂]
聚碳酸酯系树脂发泡成形体(下文中可以简称为发泡成形体)包含含有聚碳酸酯系树脂作为基材树脂的多个发泡颗粒,并且可以包含具有特定的结构和特定的沸点的增塑剂。所包含的增塑剂可以提供具有良好的熔合性的发泡成形体。即使聚碳酸酯系树脂在如上所述的特定的位置处不具有显示特定的分子量的峰,也可以显示熔合性的提高效果。通过具有所述峰,可以进一步增强提高效果。
增塑剂具有使聚碳酸酯系树脂增塑化的作用,并且优选具有2个以上的酯键、分子量为200~600且沸点为250~500℃。
在其中酯键的数量为1的情况下,其与聚碳酸酯系树脂的亲和性会是小的,并且熔合性的提高效果在一些情况下会是不足的。酯键的数量的上限为提供200~600的分子量和250~500℃的沸点的这样的值。酯键的数量优选为2~4。
在其中分子量小于200的情况下,会促进其向树脂内部的浸渍,从而提供过强的增塑化效果,并且发泡成形体在一些情况下在发泡和成形时会容易收缩。在其中分子量大于600的情况下,增塑剂的粘度会增加,并且,在一些情况下,增塑剂会难以处理并会难以提供期望的增塑化效果。分子量更优选为220~500,并且分子量进一步优选为230~450。
在其中沸点低于250℃的情况下,增塑剂在成形加工时会容易耗散,由此在一些情况下不能提供期望的效果。在其中沸点高于500℃的情况下,增塑剂在一些情况下会具有高的粘度,并且,在一些情况下,会难以处理并会难以提供期望的增塑化效果。沸点的值可以为250℃、270℃、280℃、300℃、350℃、400℃、450℃或500℃。沸点更优选为270~500℃,并且沸点进一步优选为280~450℃。
对增塑剂不特别限定,只要增塑剂具有前述结构和沸点即可,并且可以选自例如脂肪族多元羧酸与脂肪族一元醇的酯、和脂肪族多元醇与脂肪族一元羧酸的酯,并且具有2~4个酯键的化合物是优选的。增塑剂可以单独地或以其两种以上的混合物使用。
脂肪族多元羧酸的实例包括:脂肪族二元羧酸,如草酸、丙二酸、琥珀酸、戊二酸、己二酸、庚二酸、辛二酸、壬二酸和癸二酸;脂肪族三元羧酸,如丙烷三羧酸、丁烷三羧酸和戊烷三羧酸;和脂肪族四元羧酸,如丁烷四羧酸、戊烷四羧酸和己烷四羧酸。脂肪族一元醇的实例包括甲醇、乙醇、丙醇、丁醇、戊醇、己醇、庚醇、辛醇、壬醇和癸醇。
脂肪族多元醇的实例包括:脂肪族二元醇,如乙二醇、丙二醇、丁二醇、戊二醇和己二醇;脂肪族三元醇,如甘油和丁三醇;和脂肪族四元醇,如赤藓醇和季戊四醇。
脂肪族一元羧酸的实例包括:甲酸、乙酸、丙酸、丁酸、戊酸、月桂酸、肉豆蔻酸、棕榈酸、硬脂酸、油酸、山嵛酸和椰子油脂肪酸。
示例为脂肪族多元羧酸、脂肪族一元醇、脂肪族多元醇和脂肪族一元羧酸的前述化合物包括对于羧基和羟基可以被取代的位置的所有变形,并且包括对于烃基(烷基和链烷二基(alkanediylgroup)等)的可能的结构异构体。
特别优选的增塑剂的实例包括己二酸二异丁酯和甘油二乙酸单月桂酸酯。
其它增塑剂的实例包括例如甲醇、乙醇、异丙醇和甘油等多元醇系溶剂以及例如甲苯和二甲苯等烃系溶剂,但是难以通过其它增塑剂的单独使用来期望充分的增塑化。在其中使用其它增塑剂的情况下,优选通过混合具有2个以上的酯键、分子量为200~600且沸点为250~500℃的增塑剂来使用其它增塑剂。其它增塑剂的含量相对于增塑剂的总量优选为50重量%以下。可以不包含其它增塑剂。
增塑剂可以存在于构成发泡成形体的发泡颗粒中,可以存在于发泡颗粒的表面上,并且可以存在于发泡颗粒中及其表面二者。从提高熔合性的观点,增塑剂优选至少存在于发泡颗粒的表面上。
用于将增塑剂附着等至发泡颗粒的方法可以为任意已知的方法。其实例包括将增塑剂吹送至发泡颗粒上的方法、将发泡颗粒浸渍在增塑剂中的方法、和在搅拌下将增塑剂滴加至发泡颗粒上的方法。在将发泡颗粒浸渍在增塑剂中的方法中,可以将发泡颗粒预先填充在例如网状模具或冲孔板(punchingplate)等多孔性的模具内,并且将模具浸渍在填充有增塑剂的容器中。可以根据所使用的增塑剂的种类和使用量以及发泡颗粒的量适当地改变这些方法。
增塑剂可以以原液的形式附着,或可以在用例如水和醇等溶剂稀释之后附着。稀释增加液体的量并且降低粘度,由此,即使增塑剂量是小的,也可以使增塑剂均一地附着。然而,增塑剂的过度稀释会由于过低的浓度而不能提供其效果,并且会由于增塑剂向发泡颗粒内部的渗透而引起发泡颗粒的收缩。因此,根据所使用的增塑剂的种类和使用量,可以适当地改变稀释程度。
[发泡成形体的形状]
发泡成形体包含多个发泡颗粒。发泡颗粒通过其表面彼此熔合。可以通过增塑剂的存在来提供熔合性提高的发泡成形体。
发泡成形体可以具有各种密度。密度优选为0.4g/cm3以下。密度的值可以为0.01g/cm3、0.012g/cm3、0.04g/cm3、0.06g/cm3、0.12g/cm3、0.3g/cm3或0.4g/cm3。密度更优选为0.12~0.010g/cm3,并且进一步优选0.08~0.012g/cm3。
发泡成形体可以根据用途具有各种形状而没有特别限定。例如,根据例如建筑材料(土木工程材料和住宅建筑材料等)、汽车结构部件、风车等的结构部件、头盔等的结构部件、包装材料、和作为复合部件的frp的芯材等用途,发泡成形体可以具有各种形状。
[发泡成形体的制造方法]
可以例如通过对发泡颗粒赋予内压、然后使发泡颗粒进行成形步骤来获得发泡成形体。在该情况下,本发明人发现,聚碳酸酯系树脂(a)~(c)具有来自发泡性颗粒的大的发泡力。因此,聚碳酸酯系树脂(a)~(c)不仅适合于其中从发泡性颗粒经由发泡颗粒提供发泡成形体的一般的成形,而且还适合于其中从发泡性颗粒直接提供发泡成形体的所谓的原粒成形。
在制造发泡成形体之前,优选用发泡剂浸渍发泡颗粒内部从而赋予发泡力(二次发泡力)。
浸渍方法的实例包括其中通过将发泡剂压入来浸渍在搅拌下分散在水系中的树脂颗粒的湿式浸渍法、和其中将发泡颗粒投入可密闭的容器中并且通过将发泡剂压入来浸渍的实质上不使用水的干式浸渍法(气相浸渍法)。能够在不使用水的情况下进行浸渍的干式浸渍法是特别优选的。对用发泡剂浸渍树脂颗粒的浸渍压力、浸渍时间和浸渍温度不特别限定。
所使用的发泡剂可以为用于发泡颗粒制造时的发泡剂。在发泡剂中,优选使用无机发泡剂。特别地,优选使用氮气、空气、非活性气体(如氦气和氩气)和二氧化碳气体中的任一种、或其两种以上的组合。
期望用于赋予内压的压力为在不使发泡颗粒塌陷并且可以赋予发泡力的这样的范围内的压力。压力优选为0.1~4mpa,并且更优选0.3~3mpa。
浸渍时间优选为0.5~200小时。在其中浸渍时间小于0.5小时的情况下,用来浸渍发泡颗粒的发泡剂的量会太少,由此在成形时难以提供必要的二次发泡力。在其中浸渍时间长于200小时的情况下,生产性在一些情况下会劣化。浸渍时间更优选为1~100小时。
浸渍温度优选为0~60℃。在其中浸渍温度低于0℃的情况下,在一些情况下在期望的时间内会难以获得充分的二次发泡力。在其中浸渍温度高于60℃的情况下,在一些情况下在期望的时间内会难以获得充分的二次发泡力。浸渍温度更优选为5~50℃。
可以将赋予有内压的发泡颗粒供给至在发泡成形机的成形模具内形成的成形空间,然后可以将加热介质导入至其中,从而进行提供期望的发泡成形体的模具内成形。所使用的发泡成形机可以为用于从聚苯乙烯系树脂制的发泡颗粒制造发泡成形体的eps成形机、或用于从聚丙烯系树脂制的发泡颗粒制造发泡成形体的高压成形机等。要求加热介质能够在短时间内赋予高的能量,并且加热介质优选为水蒸气。
水蒸气的压力优选为0.2~0.5mpa。加热时间优选为10~300秒,并且更优选60~200秒。加热时间可以为10~90秒,并且也可以为20~80秒。
除了可以通过使用具有前述特定的气泡密度x的发泡颗粒控制气泡密度x以外,可以通过在发泡成形体的制造过程中控制浸渍条件(浸渍温度、浸渍时间和浸渍压力)和一次发泡条件(发泡压力和发泡时间)来使气泡密度x增大。可以通过控制浸渍条件(浸渍温度、浸渍时间和浸渍压力)和一次发泡条件(发泡压力和发泡时间)来使气泡密度x减小。
实施例
将参考实施例具体地描述本发明,但是本发明不限于此。用于实施例中的各种物性的测量方法在以下示出。
[通过反应热解gc/ms法的测量]
从发泡颗粒或发泡成形体取0.05~0.3mg的试样,并且在445℃的热解温度下在pyrofoil中称量。将5μl的包含20重量%的四甲基氢氧化铵五水合物(tmah,反应试剂)的甲醇溶液滴加至试样中,然后将试样干燥并且用pyrofoil包裹,从而生产热解用试样。
使用居里点热解仪(curiepointpyrolyzer)jhp-5型(由japananalyticalindustryco.,ltd.制造)和气相色谱质谱仪jms-ax505h(由jeolltd.制造:gc(hp-5890ii))作为测量设备在以下测量条件下对热解用试样进行热解测量,并且由此获得gc/ms图(总离子色谱图(tic))。在gc/ms图中,横轴示出保留时间(单位:分钟),并且纵轴示出强度(丰度(abundance))。
柱:zb-5(0.25μm×0.25mmφ×30m,由phenomenexinc.制造)
测量条件:pyrofoil(加热至445℃5秒)、炉温(280℃)、针温(250℃)、柱温(50℃(3分钟)→10℃/分→280℃→40℃/分→320℃(3分钟))、测量时间(30分钟)、载气(he)、he流量(34ml/分)、注入口温度(300℃)、sep温度(280℃)、rsv温度(80℃)、离子化电流(100μa)、cd电压(-10kv)
在所得gc/ms图中,以最大强度峰的保留时间为基准,在-15分钟以内的范围内的保留时间确认到在通过电子轰击离子化(ei)法的碎片离子的分子量中具有最大强度的分子量为145(或210)~230的峰,以相同的方式在+10分钟以内的范围内的保留时间确认到分子量为320~350的峰,并且以相同的方式在5分钟以内的范围内的保留时间确认到在通过电子轰击离子化(ei)法的碎片离子分子量中具有最大强度的分子量为290~320的峰。
通过质谱确认碎片离子的分子量,其中质谱显示以最大强度峰的保留时间为基准在-15分钟以内的范围内的峰、在+10分钟以内的范围内的峰、和在5分钟以内的范围内的峰,并且确认碎片离子的分子量。确认最大强度峰为来自碎片离子的双酚a甲基化组分。
可以通过检索对于所得质谱配备的库来鉴定结构。所述库的实例包括nist(质谱数据库)。
在所得gc/ms图中,对于最大强度峰、源自145(或210)~230的分子量的峰、源自320~350的分子量的峰、和源自290~320的分子量的峰中的每一个获得峰面积(tic峰面积)。根据下式(1)、(2)和(3)从所得峰面积获得峰面积比。
式(1):
(源自145(或210)~230的分子量的峰面积比)=(源自145(或210)~230的分子量的峰面积)/(源自双酚a甲基化物的成分的峰面积)
式(2):
(源自320~350的分子量的峰面积比)=(源自320~350的分子量的峰面积)/(源自双酚a甲基化物的成分的峰面积)
式(3):
(源自290~320的分子量的峰面积比)=(源自290~320的分子量的峰面积)/(源自双酚a甲基化物的成分的峰面积)
[mfr的测量]
熔体流动速率(mfr)使用由toyoseikiseisaku-sho,ltd.制造的"semi-automeltindexer2a"来测量,并且根据jisk7210:1999"塑料-热塑性塑料的熔体流动速率(mfr)和熔体体积流动速率(mvr)的试验方法"的b法中记载的(b)测量活塞在规定的距离内移动的时间的方法来测量。具体地,测量条件为:试样3~8g、预热270秒、负荷保持30秒、试验温度300℃、试验负荷11.77n、和活塞移动距离(间隔)25mm。试样的试验次数为3次,并且将其平均值指定为熔体流动速率(g/10min)的值。将测量试样用真空干燥机在120℃和100kpa的减压条件下干燥5小时。
[平均粒径的测量]
平均粒径为由d50表示的值。
具体地,使用转动锤击式振筛机(由sievefactoryiidaco.,ltd.制造)用开口为26.5mm、22.4mm、19.0mm、16.0mm、13.2mm、11.20mm、9.50mm、8.80mm、6.70mm、5.66mm、4.76mm、4.00mm、3.35mm、2.80mm、2.36mm、2.00mm、1.70mm、1.40mm、1.18mm、1.00mm、0.85mm、0.71mm、0.60mm、0.50mm、0.425mm、0.355mm、0.300mm、0.250mm、0.212mm和0.180mm的jis标准筛(jisz8801:2006)将约25g的试样分级10分钟,并且测量筛网上的试样重量。从由此获得的结果制成累积重量分布曲线,并且将在累积重量为50%处的粒径(中值粒径)指定为平均粒径。
[发泡颗粒的平均气泡直径的测量]
收集通过一次发泡获得的发泡颗粒。沿任意的方向切割发泡颗粒,并且用扫描型电子显微镜("s-3400n",由hitachi,ltd.制造)放大10~200倍来拍摄切出的截面的显微照片。在a4纸上打印由此拍摄的图像,选择10个任意的气泡,并且计算平均值。将该数值指定为平均气泡直径。
[发泡成形体的平均气泡直径的测量]
从长度400mm×宽度300mm×厚度30mm的成形体中央部切出长度50mm×宽度50mm×厚度30mm的片,并且用扫描型电子显微镜("s-3400n",由hitachi,ltd.制造)放大10~200倍拍摄沿切出的成形体片的厚度方向的横截面的显微照片。在a4纸上打印由此拍摄的图像,选择10个任意的气泡,并且计算平均值。将该数值指定为平均气泡直径。
[发泡颗粒的体积密度和表观密度的测量]
将约1,000cm3的发泡颗粒填充在量筒内至刻度为1,000cm3。在沿水平方向目视量筒时,在至少一个发泡颗粒达到刻度为1,000cm3的时间点,完成发泡颗粒在量筒内的填充。随后,将填充在量筒内的发泡颗粒的重量称量至小数点后2位的有效数字,并且其质量由wg示出。通过下式获得发泡颗粒的体积密度。
体积密度(g/cm3)=w/1000
体积发泡倍率为通过将体积密度的倒数乘以聚碳酸酯系树脂的密度(g/cm3)获得的值。
聚碳酸酯系树脂的密度可以通过iso1183:2004中规定的方法来测量。
通过下式获得表观密度。
表观密度(kg/m3)=(w×1000)/1000×(0.01)3
体积发泡倍率为通过将体积密度的倒数乘以聚碳酸酯系树脂的密度(kg/m3)获得的值。
[发泡成形体的密度的测量]
将从发泡成形体(成形后、在40℃下干燥20小时以上)切出的试验片(例如,75×300×30mm)的质量(a)和体积(b)各自测量至3个以上的有效数字,并且由(a)/(b)获得发泡成形体的密度(g/cm3或kg/m3)。
发泡倍率为通过将密度的倒数乘以聚碳酸酯系树脂的密度(g/cm3或kg/m3)获得的值。
[实施例1a~1c的发泡性的评价]
将其中一次发泡颗粒的体积发泡倍率达到20倍以上的情况评价为◎,将其中体积发泡倍率达到15倍以上的情况评价为○。
[成形性的评价]
对于所得发泡成形体,将300×400×30(高度)mm的发泡成形体在300×400mm的表面水平朝上的状态下,在距离其长边端20mm的部分保持,并且将其中成形体保持其形状而不断裂的情况评价为○。
[成形循环的评价]
通过以下标准来评价从成形时的加热完成至当发泡成形体的每单位面积的发泡压力变为0kgf/cm2的时刻的真空冷却时间。
○:长时间(41秒以上)
△:中程度(21~40秒)
×:短时间(20秒以下)
直至发泡成形体的每单位面积的发泡压力在加热之后立即变为0kgf/cm2的时间长意味着发泡颗粒具有高的发泡力,结果,发泡颗粒之间的熔合性是高的。
[强度的评价]
使1kg的铁球从30cm的高度落在发泡成形体上,然后通过以下标准来评价发泡成形体的状态。
○:发泡成形体表面凹陷,但是成形体不断裂。
△:成形体表面和内部开裂,但是成形体不断裂。
×:成形体断裂。
[外观的评价]
以以下方式评价外观。具体地,从发泡成形体任意地切出具有50mm×50mm的表皮的试验片,并且将试验片表面(表皮面)的颗粒间的个数计数。本文中提及的颗粒间意指3个以上的发泡颗粒彼此接触的接触点。随后,将颗粒间的针孔(pinhole)(凹陷)的个数计数。根据下式由颗粒间的个数和针孔的个数计算发泡成形体的伸长率(elongation)。本文中提及的针孔(凹陷)意指表面从在发泡成形体制造期间与模具接触的表面凹陷2mm以上的部分、或表面具有宽度为2mm以上(其为与模具接触的表面侧的宽度)的凹陷的部分。
发泡成形体的伸长率=(1-(颗粒间针孔个数)/(总颗粒间个数))×5
通过以下标准评价由此计算的伸长率的值。
外观○:4个以上(表面是光滑的。发泡颗粒彼此粘接,并且难以确认发泡颗粒的形状。)
外观×:小于4个(表面具有凹凸。确认发泡颗粒的形状。成形体是多孔状的。颗粒的形状保持。)
[实施例1d~3d中的成形性的评价]
借助以下标准通过二次发泡性来评价成形性。通过下式计算二次发泡性。二次发泡颗粒的发泡倍率意指通过用在0.34mpa下的水蒸气加热在完成内压赋予步骤之后的一次发泡颗粒30秒而显示的二次发泡颗粒的发泡倍率。
二次发泡倍率(倍)=(二次发泡颗粒的发泡倍率)/(一次发泡颗粒的发泡倍率)
○:2倍以上。
△:1.5倍~2倍。
×:1.5倍以下。
[熔合率的测量]
用切刀在发泡成形体上切出深度约为1mm的狭缝。此后,用手或锤子沿着狭缝将发泡成形体分成两片。对于暴露在断裂面上的任意的20个发泡颗粒,将在颗粒内断裂的颗粒的个数(a)计数。将通过将计数值代入式(a)×100/20而获得的值指定为熔合率(%)。熔合率为50%以上评价为○,并且熔合率小于50%评价为×。
[增塑剂量的测量]
(a)rikemalpl-012和vinycizer40
测量所述量作为甘油二乙酸单月桂酸酯或单月桂酸二异丁酯的量。
(1)萃取方法
称量约1.0g的冷冻粉碎的试样并将其直接收集至槽容器(cellcontainer)中,并且通过使用ase-350(由dionexcorporation制造)高速溶剂萃取器在以下条件下萃取。
·萃取压力:10.5mpa
·升温时间:5分钟
·静置时间:10分钟
·萃取温度:110℃
·吹扫时间:90秒
·循环次数:3次
·冲洗量:10%
·萃取溶剂:甲醇
·所需时间:45分钟
(2)rikemalpl-012的测量方法
将高速溶剂萃取液转移至容量为50ml的容量瓶中,并且在用甲醇定容后,用非水系0.20μmchromatodisc过滤,并且通过hplc测定滤液。测定条件如下,并且由从色谱图获得的标准峰面积用校准曲线获得试样溶液中的rikemalpl-012的浓度,由此计算其含量。使用校准曲线用标准样品(rikemalpl-012,由rikenvitaminco.,ltd.制造)。
(hplc测量条件)
设备:液相色谱设备,lc-10avp(由shimadzucorporation制造)
柱:tskgelods-80tsqa(4.6×150mm),由tosohcorporation制造
柱温:40℃
流动相:甲醇
流动相流量:0.7ml/分
泵温度:室温
测量时间:10分钟
注入量:50μl
检测器:蒸发光散射检测器elsd-2000(由altechassociates,inc.制造)
(检测器设定条件)
漂移管(drifttube)温度:60℃
气流:1.6ml/分
增益:1(撞击器(impactor):关)
(3)vinycizer40的测量方法
将20μl(1,000ppm)的芘作为内标液放入2ml容量瓶中,并且在用高速溶剂萃取的萃取液定容之后,借助gc/ms在以下条件下分析。
基于所得色谱图中的己二酸二异丁酯的峰面积相对于作为内标物质的芘的峰面积的相对灵敏度预先制成的己二酸二异丁酯(6.15ppm、24.6ppm和61.5ppm)的校准曲线,计算己二酸二异丁酯的浓度。此外,根据下式由试样重量和萃取液量计算含量(重量%)。
己二酸二异丁酯量(重量%)=试验液中浓度(μg/ml)×萃取液量50(ml)/试样重量(g)/10,000
(gc/ms测量条件)
测量设备:气相色谱-质谱仪qp2010se,由shimadzucorporation制造
柱:zb-5ms(0.25μm×0.25mmφ×30m,由phenomenexinc.制造)
gc炉升温条件:初期温度70℃(保持1分钟)
第一阶段升温速度:15℃/分(高达260℃,不保持)
第二阶段升温速度:10℃/分(高达300℃)
最终温度:300℃(保持3分钟)
载气:氦气
总流量·柱流量:52ml/分·1.02ml/分
柱入口压:74.9kpa
检测器:1.00kv
注入口温度:300℃
接口温度:260℃
离子源温度:260℃
分流比:1/50(内标法)
试验液注入:2μl(使用自动进样器)
测量模式:sim法(m/z=129、185、200、202)
内标液:芘
(b)其它增塑剂的测量
有必要根据增塑剂的种类适当地改变从试样萃取增塑剂的萃取条件和测量方法,用于确认增塑剂的残留量。例如,使用与rikemalpl-012类似的高速溶剂萃取器的方法适合于具有高沸点的未知物质的定量。此外,所选择的测量方法还可以为hplc和gc/ms等。可以通过ir和nmr等来进行未知物质的化合物的鉴定。优选通过用借助未知物质的鉴定制成的校准曲线确定浓度来进行残留量的确认。
以下测量方法示出对于已知物质的借助gc/ms法的分析方法。
测量试样的测量方法:将试样切成约2毫米的立方体,并且在hs小瓶中精确地称量约0.5g,立即在所述hs小瓶上放置隔膜和铝帽并且用铝帽紧固器固定。将小瓶在110℃恒温槽中加温约30分钟,并且将0.5ml的试验小瓶内的气相直接注入至gc注入口,用于测量增塑剂量。
标准的制备方法:将5μl各标准液1,000ppm甲醇溶液注入hs小瓶,立即在其上放置隔膜和铝帽并且用铝帽紧固器固定。将小瓶在110℃恒温槽中加温约30分钟,并且将0.5ml的试验小瓶内的气相直接注入至gc注入口用于测量。
测量条件如下,并且从校准曲线获得浓度。
(gc/ms测量条件)
测量设备:气相色谱-质谱仪qp2010se,由shimadzucorporation制造
柱:zb-5ms(0.25μm×0.25mmφ×30m,由phenomenexinc.制造)
gc炉升温条件:初期温度70℃(保持1分钟)
第一阶段升温速度:15℃/分(高达260℃,不保持)
第二阶段升温速度:10℃/分(高达300℃)
最终温度:300℃(保持3分钟)
载气:氦气
总流量·柱流量:52ml/分·1.02ml/分
柱入口压:74.9kpa
检测器:1.00kv
注入口温度:300℃
接口温度:260℃
离子源温度:260℃
分流比:1/50(内标法)
试验液注入:2μl(使用自动进样器)
测量模式:sim法(m/z=129、185、200、202)
内标液:芘
<实施例1a>
(浸渍步骤)
将100重量份(1,000g)的作为聚碳酸酯系树脂的lexan153(由sabic制造,密度:1.2g/cm3,mfr:4g/10min,平均粒径:3mm)投入可密闭的10l的压力容器中,并且将压力容器内的压力用二氧化碳气体升压至表压为4mpa并在室温(约20℃)的环境下保持24小时,从而提供发泡性颗粒。
(发泡步骤)
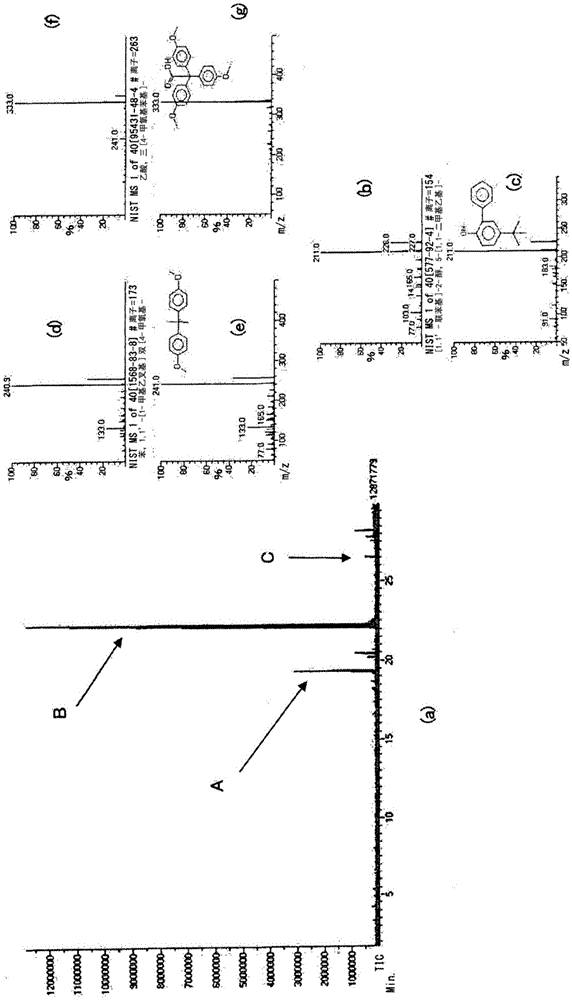
在完成浸渍之后,使压力容器内的二氧化碳气体缓慢地释放,并且将内部的发泡性颗粒取出。立即将0.3重量份(3g)的作为结合抑制剂的碳酸钙与100重量份(1,000g)的发泡性颗粒混合。此后,将发泡性颗粒投入装配有搅拌机的高压发泡机中,并且在搅拌下用0.34mpa的水蒸气发泡,从而提供体积发泡倍率为18倍(体积密度:0.067g/cm3)的发泡颗粒(一次发泡颗粒)。通过借助反应热解gc/ms测量所得发泡颗粒而获得的gc/ms图在图1(a)中示出。在图1(a)中,a表示源自210~230的分子量的峰,b表示显示源自双酚a的组分的最大峰,并且c表示源自320~350的分子量的峰。源自gc/ms图的质谱在图1(b)~1(g)中示出。图1(b)对应于源自210~230的分子量的峰,图1(d)对应于显示源自双酚a的组分的最大峰,并且图1(f)对应于源自320~350的分子量的峰。图1(c)示出通过检索对于图1(b)的库获得的结果,图1(e)示出通过检索对于图1(d)的库获得的结果,并且图1(g)示出通过检索对于图1(f)的库获得的结果。
(第二浸渍步骤:内压赋予步骤)
将所得发泡颗粒的表面用0.01n盐酸洗涤并且干燥,然后将发泡颗粒投入10l的压力容器中,然后密闭。将密闭的压力容器内的压力用氮气升压至表压为1mpa并且使其静置24小时,从而赋予内压。
(成形步骤)
在赋予内压之后,使压力容器内的氮气缓慢地释放,并且将发泡颗粒取出并立即用高压成形机进行发泡成形。将发泡颗粒填充在内部尺寸为长度400mm×宽度300mm×厚度30mm的成形用模具内,并且通过导入0.30~0.35mpa的水蒸气50秒来加热,接着冷却,从而提供发泡倍率为18倍(密度:0.067g/cm3)的发泡成形体。将所得发泡成形体在30℃的干燥室中干燥约8小时。通过借助反应热解gc/ms测量发泡成形体而获得的结果与图1(a)~1(g)基本上相同。
<实施例2a>
除了使用由bayerag制造的makrolonwb1439(密度:1.2g/cm3,mfr:3g/10min,平均粒径:3mm)作为聚碳酸酯系树脂以外,以与实施例1a中相同的方式获得体积发泡倍率为24倍的发泡颗粒和发泡倍率为24倍的发泡成形体。通过借助反应热解gc/ms测量所得发泡颗粒而获得的gc/ms图在图2(a)中示出。在图2(a)中,a表示源自145~165的分子量的峰,b表示显示源自双酚a的组分的最大峰,并且c表示源自320~350的分子量的峰。源自gc/ms图的质谱在图2(b)~2(g)中示出。图2(b)对应于源自145~165的分子量的峰,图2(d)对应于显示源自双酚a的组分的最大峰,并且图2(f)对应于源自320~350的分子量的峰。图2(c)示出通过检索对于图2(b)的库获得的结果,图2(e)示出通过检索对于图2(d)的库获得的结果,并且图2(g)示出通过检索对于图2(f)的库获得的结果。通过借助反应热解gc/ms测量发泡成形体而获得的结果与图2(a)~2(g)基本上相同。
<实施例3a>
除了使用由teijinltd.制造的panlitex0730(密度:1.2g/cm3,mfr:3.5g/10min,平均粒径:3mm)作为聚碳酸酯系树脂以外,以与实施例1a中相同的方式获得体积发泡倍率为17倍的发泡颗粒和发泡倍率为17倍的发泡成形体。通过借助反应热解gc/ms测量所得发泡颗粒而获得的gc/ms图在图3(a)中示出。在图3(a)中,a表示源自145~165的分子量的峰,b表示显示源自双酚a的组分的最大峰,并且c表示源自320~350的分子量的峰。源自gc/ms图的质谱在图3(b)~3(g)中示出。图3(b)对应于源自145~165的分子量的峰,图3(d)对应于显示源自双酚a的组分的最大峰,并且图3(f)对应于源自320~350的分子量的峰。图3(c)示出通过检索对于图3(b)的库获得的结果,图3(e)示出通过检索对于图3(d)的库获得的结果,并且图3(g)示出通过检索对于图3(f)的库获得的结果。通过借助反应热解gc/ms测量发泡成形体而获得的结果与图3(a)~3(g)基本上相同。
[表1]
从表1理解的是,如下发泡成形体在成形性和发泡性(外观)方面良好:所述发泡成形体具有以双酚a最大峰的保留时间为基准在-15分钟以内的范围内的保留时间确认到的源自145~230的分子量的峰和在+10分钟以内的范围内的保留时间确认到的源自320~350的分子量的峰。
<实施例1b>
(浸渍步骤)
将100重量份(1,000g)的作为聚碳酸酯系树脂的lexan153(由sabic制造,密度:1.2g/cm3,mfr:4g/10min,平均粒径:3mm)投入可密闭的10l的压力容器中,并且将压力容器内的压力用二氧化碳气体升压至表压为4mpa并在室温(约20℃)的环境下保持24小时,从而提供发泡性颗粒。
(发泡步骤)
在完成浸渍之后,将压力容器内的二氧化碳气体缓慢地释放,并且将内部的发泡性颗粒取出。立即将0.3重量份(3g)的作为结合抑制剂的碳酸钙与100重量份(1,000g)的发泡性颗粒混合。此后,将发泡性颗粒投入装配有搅拌机的高压发泡机中,并且在搅拌下用0.34mpa的水蒸气发泡,从而提供体积发泡倍率为18倍(体积密度:0.067g/cm3)的发泡颗粒(一次发泡颗粒)。通过借助反应热解gc/ms测量所得发泡颗粒而获得的gc/ms图在图1(a)中示出。在图1(a)中,a表示源自210~230的分子量的峰,并且b表示显示源自双酚a的组分的最大峰。源自gc/ms图的质谱在图1(b)~1(e)中示出。图1(b)对应于源自210~230的分子量的峰,并且图1(d)对应于显示源自双酚a的组分的最大峰。图1(c)示出通过检索对于图1(b)的库获得的结果,并且图1(e)示出通过检索对于图1(d)的库获得的结果。
(第二浸渍步骤:内压赋予步骤)
将所得发泡颗粒的表面用0.01n盐酸洗涤并且干燥,然后将发泡颗粒投入10l的压力容器中,然后密闭。将密闭的压力容器内的压力用氮气升压至表压为1mpa并且使其静置24小时,从而赋予内压。
(成形步骤)
在赋予内压之后,使压力容器内的氮气缓慢地释放,并且将发泡颗粒取出并立即用高压成形机进行发泡成形。将发泡颗粒填充在内部尺寸为长度400mm×宽度300mm×厚度30mm的成形用模具内,并且通过导入0.30~0.35mpa的水蒸气50秒来加热、接着冷却,从而提供发泡倍率为18倍(密度:0.067g/cm3)的发泡成形体。将所得发泡成形体在30℃的干燥室中干燥约8小时。通过借助反应热解gc/ms测量发泡成形体而获得的结果与图1(a)~1(e)基本上相同。
<实施例2b>
除了将聚碳酸酯系树脂改变为lexan131(由sabic制造,密度:1.2g/cm3,mfr:3.5g/10min)以外,以与实施例1b中相同的方式获得发泡颗粒(发泡倍率:16倍,体积密度:0.075g/cm3)。
通过借助反应热解gc/mc测量发泡颗粒(一次发泡颗粒,体积发泡倍率:16倍,体积密度:0.075g/cm3)而获得的结果在图4(a)~4(e)中示出。通过借助反应热解gc/ms测量发泡成形体而获得的结果与图4(a)~4(e)基本上相同。
[表2]
从表2理解的是,具有以双酚a最大峰的保留时间为基准在5分钟以内的范围内的保留时间确认到的源自210~230的分子量的峰的发泡成形体在成形性和发泡性(外观)方面良好。
<实施例1c>
(浸渍步骤)
将100重量份(1,000g)的作为聚碳酸酯系树脂的novarexm7027u(由mitsubishiengineering-plasticscorporation制造,mfr:5g/10min,平均粒径:3mm)投入可密闭的10l的压力容器中,并且将压力容器内的压力用二氧化碳气体升压至表压为4mpa并在室温(约20℃)环境下保持24小时,从而提供发泡性颗粒。
(发泡性确认步骤)
在完成浸渍之后,使压力容器内的二氧化碳气体缓慢地释放,并且将内部的发泡性颗粒取出。立即将0.3重量份(3g)的作为结合抑制剂的碳酸钙与100重量份(1,000g)的发泡性颗粒混合。此后,将发泡性颗粒投入能够使高压的蒸气导入和排出的发泡机中,并且通过用0.30mpa的水蒸气加热120秒来发泡。所得发泡颗粒的体积发泡倍率为30倍(体积密度:0.04g/cm3)。通过借助反应热解gc/ms测量所得发泡颗粒而获得的gc/ms图在图5(a)中示出。在图5(a)中,a表示源自290~320的分子量的峰,并且b表示显示源自双酚a的组分的最大峰。源自gc/ms图的质谱在图5(b)~5(d)中示出。图5(b)对应于源自290~320的分子量的峰,并且图5(c)对应于显示源自双酚a的组分的最大峰。图5(d)示出通过检索对于图5(c)的库获得的结果。
(成形步骤)
将4.8g的发泡性颗粒投入长度40mm×宽度80mm×厚度25mm且容积为80cm3的模具中,并且通过导入0.30~0.35mpa的水蒸气120秒来加热、接着冷却,从而提供发泡倍率为20倍(密度:0.06g/cm3)的自立式(self-standing)的发泡成形体。将所得发泡成形体在30℃的干燥室中干燥约8小时。通过借助反应热解gc/ms测量发泡成形体而获得的结果与图5(a)~5(d)基本上相同。
[表3]
从表3理解的是,具有以双酚a最大峰的保留时间为基准在5分钟以内的范围内的保留时间确认到的源自290~320的分子量的峰的发泡成形体在成形性和发泡性(外观)方面良好。
<实施例1d>
(浸渍步骤)
将100重量份(1,000g)的作为聚碳酸酯系树脂的lexan153(由sabic制造,密度:1.2×103kg/m3,mfr:4g/10min,平均粒径:3mm)投入可密闭的10l的压力容器中,并且将压力容器内的压力用二氧化碳气体升压至表压为4mpa并在20℃下保持24小时,从而提供发泡性颗粒。
(发泡步骤)
在完成浸渍之后,使压力容器内的二氧化碳气体缓慢地释放,并且将内部的发泡性颗粒取出。立即将0.3重量份(3g)的作为结合抑制剂的碳酸钙与100重量份(1,000g)的发泡性颗粒混合。此后,将发泡性颗粒投入装配有搅拌机的高压发泡机中,并且在搅拌下用0.34mpa的水蒸气发泡120秒,从而提供体积发泡倍率为23倍(表观密度:52kg/m3)的发泡颗粒(平均粒径:6mm,一次发泡颗粒)。
(第二浸渍步骤:内压赋予步骤)
将所得发泡颗粒的表面用0.01n盐酸洗涤并且干燥,然后将发泡颗粒投入10l的压力容器中,然后密闭。在0~20℃下,将密闭的压力容器内的压力用氮气升压至表压为1mpa并且使其静置24小时,从而赋予内压,由此提供发泡颗粒(二次发泡颗粒)。
(成形步骤)
在赋予内压之后,使压力容器内的氮气缓慢地释放,并且将发泡颗粒取出并立即用高压成形机进行发泡成形。将发泡颗粒填充在内部尺寸为长度400mm×宽度300mm×厚度30mm的成形用模具中,通过导入0.30~0.35mpa的水蒸气60秒来加热,使其冷却1秒,然后用水冷却10秒,并且在模具内进行真空冷却,从而提供发泡倍率为16倍(密度:75kg/m3)的发泡成形体。
<实施例2d>
除了由sabic制造的lexan121r(密度:1.2×103kg/m3,mfr:15g/10min,平均粒径:3mm)用作聚碳酸酯系树脂以外,以与实施例1d中相同的方式获得体积发泡倍率为16倍(表观密度:75kg/m3)的一次发泡颗粒(平均粒径:5mm)和发泡倍率为14倍(密度:86kg/m3)的发泡成形体。
<实施例3d>
除了由bayerag制造的makrolonwb1439(密度:1.2×103kg/m3,mfr:3g/10min,平均粒径:3mm)用作聚碳酸酯系树脂以外,以与实施例1d中相同的方式获得体积发泡倍率为21倍(表观密度:57kg/m3)的一次发泡颗粒(平均粒径:6mm)和发泡倍率为20倍(密度:60kg/m3)的发泡成形体。
实施例和比较例的平均气泡直径c、聚碳酸酯系树脂的密度ρ、一次发泡颗粒的表观密度d、发泡成形体的密度d、气泡密度x、成形循环的评价、强度的评价、外观的评价、和成形性的评价在表4中示出。
实施例的发泡颗粒的切断面的整体照片和放大的照片在图6(a)~图8(b)中示出。
图6(a)、图7(a)和图8(a)分别为实施例1d、实施例2d和实施例3d的10倍的照片。
图6(b)为实施例1d的80倍的照片,图7(b)为实施例2d的150倍的照片,图8(b)为实施例3d的150倍的照片,并且图9为实施例1d的200倍的照片。
[表4]
从表4理解的是,可以通过将气泡密度x控制至特定的范围来获得在外观和熔合性方面良好的发泡成形体。
<实施例1e>
(浸渍步骤)
将100重量份(1,000g)的聚碳酸酯系树脂(由sabic制造的lexan153(密度:1.2g/cm3,mfr:4g/10min,平均粒径:3mm)投入可密闭的10l的压力容器中,并且将压力容器内的压力用二氧化碳气体升压至表压为4mpa并在室温(约25℃)的环境下保持24小时,从而提供发泡性颗粒。
(发泡步骤)
在完成浸渍之后,使压力容器内的二氧化碳气体缓慢地释放,并且将内部的发泡性颗粒取出。立即将0.3重量份(3g)的作为结合抑制剂的碳酸钙与100重量份(1,000g)的发泡性颗粒混合。此后,将发泡性颗粒投入装配有搅拌机的高压发泡机中,并且在搅拌下用0.34mpa的水蒸气发泡,从而提供0.08g/cm3的体积密度。
(第二浸渍步骤:内压赋予步骤)
将所得发泡颗粒的表面用0.01n盐酸洗涤并且干燥,然后将100重量份(500g)的发泡颗粒投入10l的压力容器中,然后密闭。将密闭的压力容器内的压力用氮气升压至表压为1mpa并且使其静置24小时,从而赋予内压。
(成形步骤)
在赋予内压之后,使压力容器内的氮气缓慢地释放,将发泡颗粒取出,并且将100重量份(500g)的发泡颗粒和1重量份(5g)的由rikenvitaminco.,ltd.生产的rikemalpl-012(甘油二乙酸单月桂酸酯)装入塑料袋中并且通过上下振荡20次来充分混合,然后用高压成形机进行发泡成形。将发泡颗粒填充在内部尺寸为长度400mm×宽度300mm×厚度30mm的成形用模具内,并且通过导入0.30~0.35mpa的水蒸气50秒来加热、接着冷却,从而提供发泡成形体。将所得发泡成形体在30℃的干燥室中干燥约8小时,并且显示密度为0.08g/cm3。
<实施例2e>
除了将增塑剂改变为vinycizer40(由kaocorporation制造,己二酸二异丁酯)和将增塑剂的添加量改变为2.5重量份(12.5g)以外,以与实施例1e中相同的方式获得发泡成形体。
所得发泡颗粒的体积密度为0.08g/cm3,并且发泡成形体的密度为0.08g/cm3。
<实施例3e>
除了将增塑剂的添加量改变为2.5重量份(12.5g)以外,以与实施例1e中相同的方式获得发泡成形体。
所得发泡颗粒的体积密度为0.08g/cm3,并且发泡成形体的密度为0.08g/cm3。
<实施例4e>
除了将增塑剂的添加量改变为5重量份(25g)以外,以与实施例1e中相同的方式获得发泡成形体。
所得发泡颗粒的体积密度为0.08g/cm3,并且发泡成形体的密度为0.08g/cm3。
[表5]
(注1)测量下限(1ppm)以下
从表中理解的是,包含具有2个以上的酯键、分子量为200~600且沸点为250~500℃的增塑剂的发泡成形体的熔合性良好。
- 还没有人留言评论。精彩留言会获得点赞!