口腔菌群多样性的分析方法及与疾病相关的菌群标志物与流程
本发明涉及生物技术检测领域,具体地,本发明提供了一种基于二代高通量测序技术的检测疾病患者体内口腔菌群多样性的技术和方法。
背景技术:
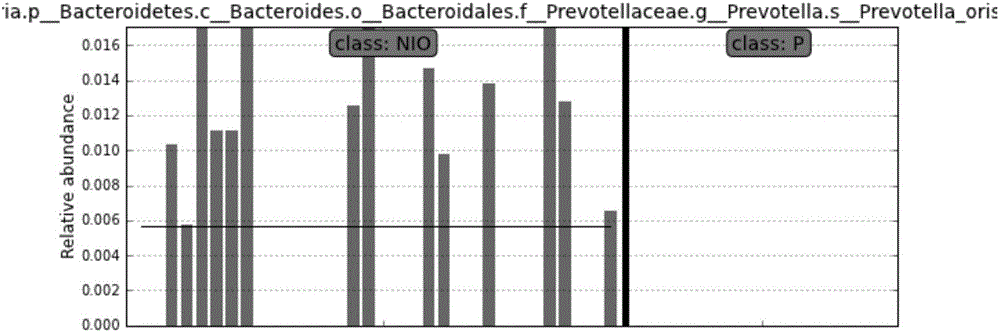
:口腔内数以亿计的微生物及其代谢产物在人体能量代谢、营养物质吸收、先天和获得性免疫、胃和口腔功能等方面发挥着重要作用,一旦宿主与口腔微生物之间共栖共生的稳态被打破,就会诱发多种人类疾病。目前已经认识到口腔微生物与人类健康和疾病的密切关系,仅仅从人类自身角度出发来研究人类疾病远远不够,必须考虑到与人类共栖共生的口腔微生物的作用。目前应用于人体口腔微生物的多样性及差异化的研究方法较多,主要有传统检测方法、构建基因文库法、遗传指纹图谱技术、分子杂交技术和DNA测序技术等方法。其中传统微生物检测方法是用各种培养基培养分离微生物,并通过革兰染色、生物化学和血清学试验等方法来确定微生物种类,通过倍比稀释、菌落计数等方法来测定微生物数量。但由于传统微生物培养技术中,菌株的富集或衰减不可避免,原始的微生态结构被改变,这会导致研究结果存在较大偏差。综上所述,应用和开发针对疾病患者口腔菌群的多样性及差异性的检测方法,能够为人们更好的了解口腔微生物以及疾病之间的相互关系的技术支撑和理论基础。技术实现要素:本发明的目的即在于提供一种针对胃癌患者口腔菌群的多样性及差异性的检测方法。本发明的再一个目的是提供一种通过设计特异性引物(人工合成的两段寡核苷酸序列,一个引物与目的基因一端的一条DNA模板链互补,另一个引物与目的基因另一端的另一条DNA模板链互补),适用于多种类疾病患者口腔菌群的多样性及差异性的检测方法。本发明的又一个目的在于为人们更好的了解口腔微生物以及疾病之间的相互关系的技术支撑和理论基础。1.在本发明中,所述引物对(上、下游引物统称为一对引物)不限于由所述序列所示的两条单链DNA分子组成的引物对,只要是能够特异性扩增所有需要检验的微生物来源的引物对均可。本发明提供了一种检测胃癌患者口腔菌群的多样性及差异性的特异性引物对,其特征在于,所述引物对是能够特异性扩增所有需要检验的微生物来源的引物对。所述引物对是由两条单链DNA分子序列组成的引物对。所述引物对的上游引物为:5’-AGAGTTTGATCMTGGCTCAG-3’;下游引物为:5’-GCTGCCTCCCGTAGGAGT-3’。所述的引物对,用于生物技术及检测领域,通过二代高通量测序技术,精准检测胃癌患者口腔菌群的多样性及差异性。2.本发明还提供一种判定待测疾病患者体内口腔菌群的多样性及差异性的方法,其特征在于包括以下步骤,(1)提供一待测唾液样品;(2)从所述唾液样品中,提取其样本DNA;(3)配置PCR反应体系,进行PCR反应;(4)对于PCR反应产物进行目标片段的检测;(5)对于有目的片段的PCR产物进行PCR产物纯化;(6)以步骤5中PCR产物为检测对象,用二代高通量测序技术进行唾液样本中的微生物种类的检测;(7)对于二代测序结果进行数据分析及处理。3.本发明所述唾液样本的采集及唾液样本DNA的提取方法为:(1)所述唾液样本的采集方法具体为:1)时间最好选择在上午的9:00-11:00之间,且在取样前的1小时之内不要进食;用清水漱口;2)漱口5分钟后,在30分钟内收集5ml唾液到一个50ml的采样管中(如果条件允许,在取样过程中,保证采养管放在冰上),提醒患者不要咳粘液;3)采样完成后,4℃,2600g,离心15min;弃上清液,将采样管的盖拧紧,做好信息记录,包括被采样者姓名、住院号及采样时间。(2)所述唾液DNA提取方法具体为:用离心后沉淀的唾液样本,选择用改进的CTAB抽提法进行样本DNA的提取。1)打开管盖,用移液枪将采样管中的沉淀转移到1.5毫升的Ep管中,12000转速,离心5分钟。2)加入300微克65℃预热的2×CTAB搅拌震荡为悬浊液。3)EP管缠绕封口膜后95℃水浴加热8分钟。4)加入300微升酚仿(取下层)混合液,振荡混匀,10000转速,离心5分钟;5)将上清液200微升移至新的1.5毫升无菌离心管中,加入3倍体积(600微升)的溶胶液,振荡混匀,分两次(每次约400微升)移入硅质柱,静置2分钟,12000转速离心30秒;6)弃液体,在硅质柱中加入约400微升的洗涤液,12000转速离心30秒;7)重复前一步骤;8)弃回收管内废液,空管离心,12000转速离心2分钟;9)将硅质柱移入新的1.5毫升无菌离心管,加入60微升TE洗脱液,(TE洗脱液需提前65℃水浴),12000转速离心30秒;10)离心管中收集的60微升液体,即为DNA模版,于-20℃冰箱保存。4.本发明所述DNA含量的判定具体为:(1)对提取出的DNA进行NanoDrop2000分光光度计检测其纯度,发现1.6<OD260/280<2.0,提取DNA浓度均>10纳克/微升。(2)所述PCR扩增反应为单重PCR反应,反应体系包含:10×buffer,正向引物和反向引物,DNA模板,dNTP,Taq酶,灭菌蒸馏水。提取的DNA为模板,用所述的引物、进行荧光定量PCR扩增。(3)所述的PCR反应的50微升的反应体系包括:试剂使用量终浓度Taq酶溶液0.8微升上游引物(10微摩)0.5微升0.1微摩下游引物(10微摩)0.5微升0.1微摩10×buffer溶液5微升DNA模板2微升dNTP溶液4微升灭菌蒸馏水37.2微升总体积15微升反应体系共:50微升。(4)所述的PCR扩增反应条件为:(5)所述的PCR反应的目标片段进行检测使用的方法是琼脂糖凝胶电泳方法。(6)所述的PCR产物纯化的具体实施步骤为:1)将PCR反应液移至一干净的1.5毫升的离心管中,加入240微升的BufferB3(从-20度冰箱取出),充分混匀。2)将混合液全部移入吸附柱,静置2分钟后,9000×g离心30秒,将滤出液再次加入吸附柱中,再次过柱子,将吸附柱放入同一收集管中。3)向吸附柱中加入500微升的WashSolution,9000×g离心30秒,弃掉收集管中的液体,将吸附柱放入同一离心管中,并重复一次。4)将空吸附柱及收集管放入离心机,9000×g离心1分钟。5)在吸附膜中央加入30微升TE,室温静置2分钟,9000×g离心1分钟。将得到的DNA置于-20度保存。5.本发明所述的二代测序的基本步骤为:(1)测序文库构建1)取DNA模板79微升(共100纳克)在1.5毫升低吸附的管子内混合试剂成分,充分混匀后在室温下孵育20分钟。2)使用试剂来纯化补平的DNA,用1.8倍体积的磁珠到样本中进行纯化。3)DNA的末端修复以及片段上链接P和A接头,在0.2毫升低吸附的管子内混合试剂成分,充分混匀后再进行PCR反应。4)使用试剂来纯化补平的DNA,用1.0倍体积的磁珠到样本中进行纯化。5)定量PCR方法定量文库浓度,用荧光定量PCR的方法对于构建好的文库进行定量,并根据定量的结果将文库稀释到100皮摩尔。(2)模板制备1)IononeTouch2仪器准备,将仪器电源打开后,进行初始化。在初始化的过程中,注意废液收集管中收集到的废液的体积。2)添加IonOneTouchOil和IonPGMOT2RecorverySolution,在添加这些试剂前应充分的将试剂颠倒混匀。3)扩增反应液准备,按照实验操作手册,将扩增反应液依次加入反应容器中,并颠倒混匀3次。4)运行IononeTouch2,在运行仪器时应注意,选择本次测序所对应的试剂盒类型:IonPGMTemplateOT2400Kit。(3)模板富集1)回收IonPGMTemplateOT2400SphereParticles,按照操作说明中注意的问题,将ISP模板进行回收。2)配置Melt-off溶液,取1.5ml离心管,按操作说明进行配置。3)准备DynabeadsMyOneStreptavidinC1Beads,将DynabeadsMyOneStreptavidinC1Beads充分震荡后,吸取13微升于1.5毫升离心管中,置于磁力架上,将酒精取出后,加入DynabeadsMyOneStreptavidinC1BeadsWashSolution130μl,然后充分震荡混匀即可。4)准备模板富集八连排,按照操作说明将8个孔内的试剂依次加入即可。5)运行ES,将八连排放入凹槽,然后在收集管里加入10微升的中和缓冲液。6)运行结束后,立即将收集管从仪器上取下,然后将试剂充分吹吸混匀。(4)上机测序1)打开PGM测序仪后,首先进行PGM水洗。在W1中加入250毫升的18MΩ的去离子水,然后按下水洗按钮即可。2)进行PGM的初始化,按照操作说明,将W1、W2、W3中加入对应的试剂,将一张干净的芯片进行初始化。在进行初始化时,应确保选择的试剂类型与本次测序相匹配。3)准备dNTP溶液,在每个50ml的离心管里加入20微升的dNTP溶液,然后换上新的sipper,旋紧。4)测序程序设定,用电脑访问测序仪所对应的服务器,然后根据本次测序所用的试剂、耗材,以及后续分析所要求的结果,进行测序程序的设定。5)芯片的检验,取一张全新的芯片,然后做芯片检测,如果通过,说明芯片没有问题,可以进行后续实验步骤。6)上样模板准备,向所有的ISP中加入ControlIonSphereParticles,作为对照。加入测序引物,进行PCR反应,使二者可以连接。在加入3微升的酶,然后室温静置5分钟即可。7)芯片处理,将做完芯片检测的芯片从测序仪上取下,将里面的液体吸出。8)芯片上样,用移液器将30微升的样本缓慢的旋入芯片内,然后在甩片机上进行进样孔朝向里-外-里三次甩片。9)运行测序程序,将上样好的芯片放在测序仪上,操作显示屏,选择之前设定好的测序程序,进行测序。6.本发明所述引物在生物技术检测领域,提供了一种基于二代高通量测序技术,检测疾病患者口腔菌群的多样性及差异性的方法。在上述方法中,所述待测样本可为胃癌患者及正常人的唾液样本。如胃癌、肾病等。本发明在二代高通量测序的基础上,设计了可以扩增多种微生物的通用引物,建立了基于二代高通量测序技术,检测疾病患者口腔菌群的多样性及差异性的方法。与常规方法相比,本发明的方法是通过PCR技术对患者体内的口腔菌群提取出来的DNA进行了V1-V2区域的扩增,对患者体内的口腔菌群的多样性及差异性进行了研究,并且利用Iontorrent平台进行测序,仅两天就可以对100个左右的样本完成测序,大大节省了时间。附图说明图1为胃癌患者组及健康人群中,差异物种在各样本中的相对丰度直方图;图2为胃癌患者组及健康人群中,不同群落间微生物的差异分析的菌群生物标志物图;图3为慢性肾脏病及健康人群中,不同群落间微生物群落差异的PCoA图;图4为慢性肾脏病及健康人群中,不同群落间微生物群落差异的菌群生物标志物图;具体实施方式在本发明的实施例1中,所述待测样本为系统疾病患者唾液样本,具体为正常人的唾液样本32例,胃癌患者的唾液样本17例。共测疾病样本49。采用上述方法,对不同患者体内的口腔菌群进行DNA的提取,并将提取的DNA进行PCR反应,对PCR反应的产物进行琼脂糖凝胶电泳,然后进行PCR产物纯化,构建文库,准备上机测序,依次进行以下步骤:1.唾液样本取样方法严格按照试验操作流程进行唾液样本的取样。2.唾液DNA的提取方法用CTAB法进行唾液DNA的提取。3.DNA含量的判定对提取出的DNA进行NanoDrop2000分光光度计检测其纯度,发现1.6<OD260/OD280<2.0,提取DNA总量均>10纳克/微升。4.细菌的特异性引物的选择根据已有的引物、引物的Tm值,设计PCR反应程序时的解链温度。5.PCR扩增反应体系严格按照试剂配比进行PCR反应体系的配置。6.PCR扩增反应程序严格按照PCR扩增反应程序进行程序的设置。7.PCR产物的特异性检测利用琼脂糖凝胶电泳检测目标片段是否在355bp左右。8.V1-V2区的特异性扩增后的PCR产物的纯化严格按照试验操作流程,进行PCR产物的纯化。9.使用QUBIT进行精确的DNA浓度定量将60-90个左右的样本进行DNA浓度的精确定量,然后根据测得的最小浓度,将所有的样本进行同一浓度的稀释。10.测序文库构建将纯化后的PCR反应产物按照实验操作流程加入EP管内,进行接头的连接,末端修复等。11.模板制备将模板制备的仪器水洗后,进行模板的制备。12.模板富集将模板富集的机器打开后,先提前运行一次。然后,将收集管内,加入中和缓冲液,运行机器。13.上机测序将富集完成后的模板,进行加接头、加测序反应引物后,进行芯片上样,然后设定测序反应程序,进行上机测序。14.利用QIIME进行生物信息学分析。(1)原始数据预处理原始数据经过去接头、低质量过滤等处理,获得高质量的Reads序列,然后合并正反向Reads,获得16SrDNAV1-V2区扩增子序列。利用FLASH软件合并正方向reads,该软件可以快速、准确的检测到正反向reads的Overlap序列,并将合并后序列输出,其准确性在99%以上。(2)合并数据长度分布及质量统计合并后数据使用FastQC软件进行指控分析,该软件从原始数据统计到数据质量、序列长度、GC偏好、冗余等信息以网页的形式展现出来,可以直观的看出数据的指控信息。(3)配置文件的准备配置文件中包含了混合扩增子的各个样本的重要信息,如样本名称、标签序列、引物序列、样本处理、采集区域、采集时间等。(4)分离样本序列根据配置文件标签序列信息分离样本,统计各个样本基本信息。(5)去除嵌合体采用denovo方式去除嵌合体序列,嵌合体检查软件为usearch61。(6)筛选操作分类单元(OperationalTaxonomicUnits,OTUs)将所有分离的高质量样本序列做聚类分析,聚类可以将一定同源性的序列聚为一类(默认同源性为97%),每一个OTU代表一个物种。(7)分类学组成分析使用RDP分类器和Greengenes参考数据集。(8)样本内多样性分析(α多样性分析)主要是稀疏曲线分析。(9)样本间多样性分析(β多样性分析)数据产生的距离矩阵可被主坐标分析(PrincipalCoordinateAnalysis,PCoA)可视化。(10)LEfSe分析LDA分值直方图、微生物群落差异分类图及差异特征的相对丰度直方图。本次实验共测疾病样本即胃癌患者的唾液样本17例,正常人的唾液样本32例。共测疾病样本49例。用数据分析软件对测序结果进行质控以及分析后,得出不同样本之间群落微生物的差异(见图1),并且对结果进行不同分类样本微生物群落差异的分析,从而找到相对应的生物标志物(见图2)。在本发明的实施例2中,所述待测样本为唾液样本,具体为正常人的唾液样本330例,慢性肾脏病病人的唾液样本166例。采用上述方法,对不同患者体内的口腔菌群进行DNA的提取,并将提取的DNA进行PCR反应,对PCR反应的产物进行琼脂糖凝胶电泳,然后进行PCR产物纯化,构建文库,准备上机测序,依次进行以下步骤:1.唾液样本取样方法严格按照试验操作流程进行唾液样本的取样。2.唾液DNA的提取方法用CTAB法进行唾液DNA的提取。3.DNA含量的判定对提取出的DNA进行NanoDrop2000分光光度计检测其纯度,发现1.6<OD260/OD280<2.0,提取DNA总量均>10纳克/微升。4.细菌的特异性引物的选择根据已有的引物、引物的Tm值,设计PCR反应程序时的解链温度。5.PCR扩增反应体系严格按照试剂配比进行PCR反应体系的配置。PCR扩增反应程序严格按照PCR扩增反应程序进行程序的设置。7.PCR产物的特异性检测利用琼脂糖凝胶电泳检测目标片段是否在355bp左右。8.V1-V2区的特异性扩增后的PCR产物的纯化严格按照试验操作流程,进行PCR产物的纯化。9.使用QUBIT进行精确的DNA浓度定量将60-90个左右的样本进行DNA浓度的精确定量,然后根据测得的最小浓度,将所有的样本进行同一浓度的稀释。10.测序文库构建将纯化后的PCR反应产物按照实验操作流程加入EP管内,进行接头的连接,末端修复等。11.模板制备将模板制备的仪器水洗后,进行模板的制备。12.模板富集将模板富集的机器打开后,先提前运行一次。然后,将收集管内,加入中和缓冲液,运行机器。13.上机测序将富集完成后的模板,进行加接头、加测序反应引物后,进行芯片上样,然后设定测序反应程序,进行上机测序。14.利用QIIME进行生物信息学分析。(1)原始数据预处理原始数据经过去接头、低质量过滤等处理,获得高质量的Reads序列,然后合并正反向Reads,获得16SrDNAV1-V2区扩增子序列。利用FLASH软件合并正方向reads,该软件可以快速、准确的检测到正反向reads的Overlap序列,并将合并后序列输出,其准确性在99%以上。(2)合并数据长度分布及质量统计合并后数据使用FastQC软件进行指控分析,该软件从原始数据统计到数据质量、序列长度、GC偏好、冗余等信息以网页的形式展现出来,可以直观的看出数据的指控信息。(3)配置文件的准备配置文件中包含了混合扩增子的各个样本的重要信息,如样本名称、标签序列、引物序列、样本处理、采集区域、采集时间等。(4)分离样本序列根据配置文件标签序列信息分离样本,统计各个样本基本信息。(5)去除嵌合体采用denovo方式去除嵌合体序列,嵌合体检查软件为usearch61。(6)筛选操作分类单元(OperationalTaxonomicUnits,OTUs)将所有分离的高质量样本序列做聚类分析,聚类可以将一定同源性的序列聚为一类(默认同源性为97%),每一个OTU代表一个物种。(7)分类学组成分析使用RDP分类器和Greengenes参考数据集。(8)样本内多样性分析(α多样性分析)主要是稀疏曲线分析。(9)样本间多样性分析(β多样性分析)数据产生的距离矩阵可被主坐标分析(PrincipalCoordinateAnalysis,PCoA)可视化。(10)LEfSe分析LDA分值直方图、微生物群落差异分类图及差异特征的相对丰度直方图。本实施案例共测样本496例,其中正常人样本330例,慢性肾脏病166例。用数据分析软件对测序结果进行质控以及分析后,得出不同样本之间群落微生物的差异(见图3),并且对结果进行不同分类样本微生物群落差异的分析,从而找到相对应的生物标志物(见图4)。当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1