DNA测序装置的控制系统及控制方法与流程
本发明涉及dna测序技术领域,具体是涉及一种基于焦磷酸测序的dna测序装置的控制系统及控制方法,属于基因检测领域。
背景技术:
一、焦磷酸测序概况
焦磷酸测序技术(pyrosequencing)是于1987年发展起来的、基于dna合成过程中释放的焦磷酸(ppi)检测的测序技术,焦磷酸测序反应在一系列酶的催化作用下的,其过程中会产生与脱氧三磷酸核苷(dntp)的聚合数目成比例的可见光,通过对可见光检测可达到测定dna序列的目的。焦磷酸测序有两种实现方法:液相焦磷酸测序法(liquidphasepyrosequencing)和固相焦磷酸测序法(solidphasepyrosequencing)。
液相焦磷酸测序是由4种酶催化的同一反应体系中的酶级联化学发光反应,其原理是:引物与模板dna退火后,在dna聚合酶(dnapolymerase)、atp硫酸化酶(atpsμlfurylase)、荧光素酶(1uciferase)和三磷酸腺苷双磷酸酶(apyrase)协同作用下,将引物dna上每一个dntp的聚合与荧光信号的释放偶联起来,通过检测荧光的释放和强度,达到实时测定dna序列的目的(马永平等,焦磷酸测序技术及其在分子生物学领域的应用[j].国外医学分子生物学分册2003,25(2):115-117)。
液相焦磷酸测序的反应体系由反应底物、待测单链、特异性测序引物和酶构成,反应底物为5’-磷酰硫酸(aps)和荧光素(1uciferin)。液相焦磷酸测序反应过程是一个向反应体系中轮流加入4种dntp参与反应的过程,每轮反应只有一种dntp参加。如果加入的dntp刚好能和dna模板的下一个碱基配对,则其在dna聚合酶的作用下被添加到测序引物的3’末端,同时释放出一个分子的焦磷酸(ppi);在atp硫酸化酶的作用下,生成的ppi和aps结合形成atp;在荧光素酶的作用下,生成的atp又和荧光素结合形成氧化荧光素,同时产生可见光。如果加入的这种dntp刚好能和dna模板的下面连续n个相同碱基匹配,根据反应方程式可知,释放出的可见光强度应是只有1个碱基匹配时的n倍,也即反应过程中释放的光强度与相匹配的碱基数目成比例关系。如果加入的dntp和dna模板的下一个碱基不匹配,则上述反应不会发生,也没有可见光的释放。未参加反应的dntp和atp在核苷酸降解酶apyrase的作用下降解。
每轮反应释放出的可见光通过微弱光检测装置转化,再被处理为数字信号,经pc软件处理即可获得一个特异的检测峰,峰值的高低应和相匹配的碱基数目成比例关系。
待上一轮反应结束后,加入另一种dntp,重复进行上述反应。最终可根据获得的光强度峰值图即可读取的待测dna序列信息。
需要注意的是:datp的降解产物脱氧单磷酸鸟苷(damp)是荧光素酶的抑制剂,随着反应的进行,其浓度会越来越高,会阻碍焦磷酸测序化学发光反应的继续进行。这也是焦磷酸测序法测序长度较短(通常为20bp~30bp)的主要原因(shendurej,eta1.advancedsequencingtechnologies:methodsandgoals,nat.revgenet.,2004,5(5):335-44)。
固相焦磷酸测序是由3种酶催化的化学发光反应,与液相焦磷酸测序相比,没有三磷酸腺苷双磷酸酶参加。固相焦磷酸测序反应过程如下:结合了引物的dna模板被固定于支撑物上并在反应过程中保持位置不变;加入一种dntp后,在dna聚合酶、atp硫酸化酶和荧光素酶协同作用下发生反应,除了没有降解反应发生,其他反应与液相焦磷酸测序完全相同;在加入下一种dntp前有一个冲洗步骤(washingstep)将上轮反应残留物完全冲走,不会有抑制性产物的堆积。
通常情况下,人们所说的焦磷酸测序法指的是液相焦磷酸测序法,因为其四酶反应系统使得焦磷酸测序可以很方便地在单管中实现。
ronaghi等利用datpαs替代datp提高焦磷酸测序的信噪比(ronaghim.eta1.real-timednasequencingusingdetectionofppirelease;anal.biochem,1996,242(1):84-89)。因为datpαs可以比datp被dna聚合酶更有效地利用,更有利于阅读富含t的区域。datpαs是两种异构体sp-datpαs和rp-datpαs的混合物,聚合酶只能利用sp-datpαs。为了得到最佳反应效率,必须在反应体系中保持最佳浓度的sp-datpαs,与此同时rp-datpαs的浓度也在增加。datpαs被apyrase降解后仍会产生荧光素酶的抑制物,因此datpαs的加入并没有提高读序能力。
gharizadeh等人对此进行了改进,他们在反应中只加入纯的sp-datpαs,而不加入无用的rp-datpαs,提高了反应的效率,大大降低了抑制产物的浓度,使得荧光素酶能够维持较长时间的活性,使焦磷酸测序法的测序长度增加到50bp~100bp,测序长度的增加也使得焦磷酸测序技术有了许多新的应用(gharizadehb.etal.,long-readpyrosequencingusingpure2’-deoxyadenosine-5’-0’-(1-thiotriphosphate)sp-isomer;analbinchem,2002.301:82-90)。
在2000年举办的第十二届基因组测序和分析会议(12thinternationalgenomesequencingandanalysisconference)上,ronaghi等人提出了一种移除抑制产物、减小稀释效应的方法,将测序长度增加到200bp。
与sanger双脱氧链终止测序法相比,焦磷酸测序法具有快速、准确、经济、实时检测的特点;它不需要凝胶电泳,也不需要对dna样品进行任何特殊形式的标记和染色,具有很高的可重复性;能实现高度的并行性和高度的自动化。
二、焦磷酸测序技术的应用进展
1在单核苷酸多态性研究中的应用
单核苷酸多态性(singlenucleotidepolymorphisms,snps)是近年来出现的第三代遗传标记,它指在基因组内特定核苷酸位置上存在两种不同的碱基,其中最少的一种在群体的频率不小于1%。snps是生物的基因组中最为常见的遗传多态型,它可以在任何一个待研究基因的内部或附近提供一系列标记;而正是这种基因组中的多态性,即基因组序列的差异构成了不同个体与群体对疾病的易感性、对药物与环境因子不同反应的遗传学基础。
snp的研究主要包括两个方面:一是snp数据库的构建,主要是发现特定种类生物基因组的全部或部分snp。二是snp功能研究,发现snp只是snp研究的第一步,snp功能的研究才是snp研究的目的。sanger测序技术已成为大规模、准确、快速发现snp的主流技术。而对数据库中已有snp进行序列验证分析和频率分析,擅长短序列测序和验证的焦磷酸测序技术是很好的选择,采用焦磷酸测序技术进行snp研究,更可以节约时间并降低消耗。
nordfors等分别采用taqman荧光定量法和焦磷酸测序技术对高达1022个样本进行snp基因分型研究,获得了相同的结果,此对照实验表明焦磷酸测序技术是进行高通量、大样本的snps的高效、高准确度的方法。wasson等利用焦磷酸测序技术来进行dna池(dnapools)的snp等位基因频率分析。rickert等采用焦磷酸测序技术对4倍体马铃薯进行基因型研究,在对94个多态性位点检测中,有76个等位基因位点可用焦磷酸测序技术鉴别,有效率达81%。蒋思文等利用焦磷酸测序技术进行了鉴别猪线粒体细胞色素b基因单倍型的工作。袁建林等利用焦磷酸测序技术进行hla-drb基因型分析研究,实验表明,将焦磷酸测序结果与hla数据库的基因序列比较后可准确进行基因分型,该方法可应用于临床器官移植的供体/受体筛查。
2在病原微生物快速鉴定中的应用
jonasson等用焦磷酸测序技术检测病原菌16srrna基因,快速鉴定出临床标本中抗生素抵抗菌。monstein等用焦磷酸测序技术成功检测幽门螺杆菌16srrna基因易变的v1和v3区序列,证明该技术可满足对临床病原菌标本的快速鉴定和分型。unnerstad等利用该技术对106株不同血清型的单核细胞增生李斯特氏菌进行了分型,利用焦磷酸测序技术在短时间内完成大量的样本测序,其并行性和高效率非常显著。gharizadeh等用此技术对67个人乳头瘤病毒样品进行了鉴定和分型,证明该技术也非常适于hpv等病原体的大规模鉴定、分型和突变的研究。程绍辉等从感染人sars病毒的vero-6细胞中提取病毒rna,采用焦磷酸测序技术对多个碱基突变位点测序和突变频率分析。通过测序分析多个可能出现突变的位点,确定了该病毒为北京流行株。
3在病因学研究中的应用
kittles等利用焦磷酸测序技术,对尼日利亚人、欧洲裔美国人和非洲裔美国人三个不同种群的cypl7基因多态性进行分析,研究非洲裔美国人的cypl7基因多态性与前列腺癌之间的关系和临床表现。研究结果表明,序列为cc的cypl7基因型的非洲裔美国人比序列为tt的cypl7基因型非洲裔美国人患前列腺癌的几率要高,证明这类人群中的碱基为c的cypl7基因多态性与前列腺癌的发病率关系密切,为高危人群。众多线索表明,位于染色体22q11的comt基因与精神分裂症的发病存在重要联系,而科学家的研究工作一直没能提出有力的证据;shifman等提出了一种有效的方法,他们利用焦磷酸测序技术,对大样本的德系犹太人群进行相关的单核苷酸多态性分析,证实精神分裂症的发生与cmot基因之间存在高度的关联。这种方法也能适用于其他疾病的基因分析研究。
4在法医鉴定中的应用
用sanger测序法对线粒体dna(mtdna)变异分析,不能实现对由含有污染物、多个个体dna等组成的mtdna混合物的精确量化分析,而andreasson等提出了一种针对mtdna混合分析的基于焦磷酸测序技术的新颖的量化方法,可以很容易地从法庭证物混合样本中快速、准确地检测出主要的和次要的mtdna成分。balitzki-korte利用焦磷酸测序技术对线粒体12srrna基因进行测序分析,在149bp长度的基因片断上进行长度为20bp的检测,通过参考数据库序列,就能够充分确定对象的生物学起源,比如说,能够确定一块皮肤组织究竟是来自失踪人员还是动物身上。
三、焦磷酸分析装置及发展
焦磷酸测序技术的应用依赖于焦磷酸分析装置的研究与开发。不论何种焦磷酸分析装置,其主要结构都应包含两个部分:反应器部分和微弱光检测部分。反应器提供反应进行的场所,微弱光检测部分负责检测反应发出的可见光。在人们对焦磷酸测序技术的研究和应用过程中,设计和使用的反应器主要可以分为3类:微量板反应器、微流控芯片反应器和微阵列芯片反应器。
而商业化的焦磷酸测序仪在国外已面世,然而国内相关仪器本身研究的报道甚少,更无相应的产品问世。国外产品的一个典型代表是pyrosequencingab公司的psq96,它是该公司2001年推出的产品,系统能够同时进行96路或者384路dna样本的独立测序,当测序长度不超过300bp时一般所用时间在1小时45分钟,准确性和可靠性达到99%,具有高通量、快速、经济的优势。psq96系统已经广泛用在基础医学研究和临床分子诊断当中。
国外仪器研究另外一个代表是美国454lifescience公司2005年推出的genomesequencer20(gs20)。它走向了有着更高技术含量的微型化方向,即利用mems技术将微滤腔作为焦磷酸测序反应的反应环境,将惊人的上百万数目的反应阵列集成进7cm×8cm的面积内,并使每个反应舱能独立同时进行测序级联反应,仪器具有的高灵敏度和分辨率的ccd能够捕捉到每个单反应舱产生的微弱荧光信号,最终能够得到每个标本dna的序列信息。gs20仅需4.5个小时即可实现高密度测序反应,经过并行计算得到每个标本的序列信息。优点是可以节省反应试剂的消耗,降低测序成本,为基因组大规模测序提供可能。
目前国内的仪器研究刚刚起步,国产化的道路漫长,面对方方面面的问题,在权衡测序系统所需要的各种硬件条件后,发现首要面对的问题和挑战是微量加样子系统的研制问题。
四、焦磷酸测序中加样系统的重要性
焦磷酸测序技术及其产品为大通量、低成本、适时、快速、直观地进行单核苷酸多态性研究和临床检验提供了非常理想的技术操作平台,是后基因组时代进行基因序列分析研究的有力工具。焦磷酸测序技术正被越来越多的研究人员接受和采用,随着国际上焦磷酸测序技术应用的兴起和商品化焦磷酸测序仪器的发展,我国的焦磷酸测序技术应用方兴未艾。但是在现阶段,国内焦磷酸测序技术的应用和推广存在几个制约因素:(1)现有的商品化焦磷酸测序仪器如psq96、gs20价格昂贵;(2)商业化的焦磷酸测序服务等待时间长且很不方便;(3)虽然目前有些实验室在进行诸如焦磷酸测序芯片等装置的研究,一些实验室有自制的、结构简单的焦磷酸测序试验装置,但国内没有面向低端的、价格便宜的、商品化的基于焦磷酸测序技术的序列检测仪器,是制约焦磷酸测序技术应用发展的关键问题。
焦磷酸测序系统是在微量环境中进行的,通常反应体系仅在50μl,所需的反应底物、dna模板以及脱氧核苷酸等试剂的量非常微小;同时,单次加样量的多少直接影响循环反应的可持续性,过大的单次加样量会使反应溶液体积迅速变大,因此造成模板浓度下降过快。由于扩散作用产生的反应延迟非线性增加,得到的微弱荧光信号在时间轴上延伸,强度在纵坐标上降低,最终导致严重缩短核酸的可测序长度。一般认为由于后续加样造成的反应体系增加如果在10%之内,对实验结果的影响是在可以接受的范围内。假如要对一段20bp长度的dna片断进行测序,那么在50μl的反应体系中,允许的单次加样量只能不超过0.3μl。
此外,除了加样精度,加样间隔的时间准确性也同样重要。只有在等时间间隔内加入单循环所需的dntp,才能使每次的残留dntp的降解程度相当,那么对下次反应造成的影响也将相等。等时间段才能给每个周期的信号提供基准,便于后续根据荧光信号强度计算核苷酸结合数目的自动化分析。
虽然国产化的加样设备已比较丰富,但是国内现有的微量加样装置都有不足。比如上海复日的加样平台,作为大型自动化样本池处理设备,能够进行标准96孔板的加样、振荡、清洗工作,但是这套系统由于喷嘴加工工艺的限制,加样微精度最小只有1μl,无法满足焦磷酸测序要求的nl级别。经过调研,限制于国内应用和制造水平,国产化的加样装置都无法满足焦磷酸测序中对加样量以及重复精度的高要求。
东南大学葛健徽等公开了一种以微弱光检测模块和微量dntp加样模块为关键模块的液相焦磷酸分析装置,其中公开了一气压控制微量dntp加样模块,可实现96路dntp溶液的同时加样,最小加样量为1.2μl,最大误差为13%;但信号噪声较大,仍需更一步改进(葛健徽等,基于焦磷酸测序的基因检测装置的研制,东南大学,硕士学位论文,2006)。
王春林等公开了一种采用压电陶瓷喷头的焦磷酸核酸测序仪中微量加样系统,该可以在步进电机驱动下,对96孔标准板样本分别进行4种dntp试剂的轮流加样,加样重复精度大于95%,单次加样最小量能达到0.lμl。上述两类较佳的焦磷酸核酸测序仪中所用的微量加样系统结构比较复杂,且加样针易堵,dntp通过不同的方式喷入测序反应液内并不与反应液接触使得与反应液混合不充分反应不完全,对dntp要求量也较高且数据易不准确;此外,拆装麻烦,成本高且不利于特殊条件下应用。
焦磷酸测序(preosequencing)技术是近年来发展起来的一种新的dna序列分析技术,它通过核苷酸和模板结合后释放的焦磷酸引发酶级联反应,促使荧光素发光并进行检测。是一个理想的遗传分析技术平台,既可进行dna序列分析,又可进行基于序列分析的单核苷酸多态性(snp)检测及等位基因频率测定等,该项技术目前已被广泛应用于医学生物等各个领域。
焦磷酸测序是由dna聚合酶(dnapolymerase)、三磷酸腺苷硫酸化酶(atpsulfurylase)、荧光素酶(luciferase)和双磷酸酶(apyrase)4种酶催化同一反应体系的酶级联化学发光反应,反应底物为5’-磷酰硫酸(adenosine5’phosphosulfate,aps)和荧光素。反应体系还包括待测序dna单链和测序引物。在每一轮测序反应中,加入1种dntp,若该dntp与模板配对,聚合酶就可以将其掺入到引物链中并释放出等摩尔数的焦磷酸基团(ppi)。硫酸化酶催化asp和ppi形成atp,后者驱动荧光素酶介导的荧光素向氧化荧光素的转化,发出与atp量成正比的可见光信号,并由pyrogramtm转化为一个峰值,其高度与反应中掺入的核苷酸数目成正比。根据加入dntp类型和荧光信号强度就可实时记录模板dna的核苷酸序列。在实验过程中用α-硫化的三磷酸腺苷(datpαs)代替三磷酸腺苷(datp)以有效地被dna聚合酶利用,而不被虫荧光素识别。由于spdatpαs可以降低datpαs降解产物的浓度,近年来,单链dna结合蛋白(singlestarnddnabindingportein,ssbp)和纯化spisomerdatpas的使用datpαs降解产物抑制双磷酸酶活性的这一问题得到较好解决,使得测序长度可达10bp,拓展了该技术在遗传学领域的应用范围。
焦磷酸测序中,通过使用阶段性互补链合成反应和化学发光反应,检测发光,从而来确定dna序列。将含有由互补链合成产生的焦磷酸和剩余的核酸底物的反应液从而进行互补链合成的反应槽移动到另外的反应槽,进行发光反应,并在反应液移动的过程中通过固定有分解剩余的核酸底物的酶的区域,将剩余的核酸底物分解后,使焦磷酸转化为atp,导入化学发光反应槽。但现有技术的弊端是必须加入大量的底物和酶进行反应,以保证反应可以完全进行,之后需要再反应掉多余的底物后清洗,这不仅增加了反应过程,增大了每一步的清洗及反应难度,还会对底物等试剂造成很严重的浪费,反应时间长,浪费人力物力,无形中降低了焦磷酸测序在市场中的推广和使用潜力。
目前应用于焦磷酸测序的仪器多为部分厂家垄断制造和销售,仪器与试剂均为配套销售,测序时成本十分昂贵,检测维修过程均需依赖于特定的技术人员,周期长成本高,反应所需的体积较大,更加大了反应的成本,检测结果不稳定,重复精度低。因此设计自主研发开发一款适用于焦磷酸测序的dna测序仪是十分有必要的。
五、dna单链分离技术概况
dna单链分离技术是生物医学领域中最常见的分离技术之一,适用于不同核酸样品的dna各种规模测序及探针设备,广泛地应用于生物学、医药学、预防医学、动植物学、农牧业、食品与卫生、能源与化工、环境监测以及医学诊断与检测等领域。此外,dna单链的吸附、提取与分离技术在水质、水源、生物材料、生物体液(如血液、血清、血浆、脑脊液、尿液、泪液、汗液、消化液、精液、分泌液、组织液、呕吐物、粪便)、组织/细胞和微生物裂解液、不同来源的蛋白、核酸等生物、化学分子以及药物等的分析检测、分离和纯化以及寡核苷酸、多肽、先导化合物和药物的合成等方面被广为应用,与人们的日常生活息息相关,在生物医药领域具有举足轻重的地位。
生物医药领域中常用的dna单链分离方法并存在的不足如下:
1.热变性或碱处理。该种方法主要是将双链pcr产物进行加热或碱处理,由于dna双链在高温或一定程度的碱性环境下氢键会断裂,使得dna变为单链。虽然该种方法原理可行,操作简单,但该种方法由于其分离率和纯度较低而逐渐不作为单链dna的纯化,多用于dna的双链分离。
2.t7逆转录法。该种方法是在一条pcr引物5’端加上t7启动子,以纯化pcr扩增产物为模板,用t7rna聚合酶体外逆转录合成单链rna(hughes,et.al.,nat.biotechnol.,2001,19:342-347)。该种方法虽然原理可行,dna单链分离率较高,且获得的dna单链纯度较高,但整个分离过程需分为两大步骤完成,操作不便,时间较长,而且需严格控制rna酶的污染,故具有一定的局限性。
3.核酸外切酶法(higuchiandochman,nucleic.acidsres.,1989,17:5865)。由于一条pcr引物被磷酸化,pcr产物在用核酸外切酶消化时,被磷酸化的引物扩增链不被切割,消化后酶被加热灭活。该种方法也需纯化pcr产物,分离程序冗长,操作亦十分不便,而且dna单链获得率依赖于外切酶的活性,不可控因素过强,实验结果的稳定性不够;因此,该种方法的推行率不广,通用性不高。
4.变性高效液相色谱法(denaturinghigh-performanceliquidchromatography,dhplc)。在部分变性的条件下,通过杂合与纯合二倍体在柱中保留时间的差异,发现dna突变。异源dna双链与同源dna双链的解链特性不同,在部分变性条件下,异源双链因有错配区的存在而更易变性,在色谱柱中的保留时间短于同源双链,故先被洗脱下来,在色谱图中表现为双峰或多峰的洗脱曲线。由于一条pcr引物被生物素标记,其pcr扩增链在dhplc时,会与另一条普通链分开(dickmanandhornby,anal.biochem.,2000,284:164-167)。该方法可以在15min内直接从双链pcr产物中获得所需的dna单链,但该种方法的实施需要配套十分昂贵的仪器,故始终难以普及。
5.磁珠捕获法。运用纳米技术对超顺磁性纳米颗粒的表面进行改良和表面修饰后,制备成超顺磁性氧化硅纳米磁珠。该磁珠能在微观界面上与核酸分子特异性地识别和高效结合。利用氧化硅纳米微球的超顺磁性,在chaotropic盐(盐酸胍、异硫氰酸胍等)和外加磁场的作用下,能从血液、动物组织、食品、病原微生物等样本中的dna和rna分离出来,然后用naoh处理得到目标单链。该种方法操作简单、用时短,整个提取流程分为四步,大多可以在36-40分钟内完成,且安全无毒,不使用传统方法中的苯、氯仿等有毒试剂,对实验操作人员的伤害减少,符合现代环保理念,磁珠与dna单链的特异性结合使得提取的dna单链纯度高、浓度大,但该种方法中使用的包被磁珠较为昂贵,且需要依靠磁力架分离,不仅分离成本较高,还不方便,故在一定程度上限制了该技术的推广。
6.不对称pcr。以上方法均需在pcr后进行额外的处理,而不对称pcr可在pcr扩增的同时制备dna单链。常规不对称pcr使用两条不等量的引物,在开始的循环里进行正常扩增。随着循环的增加,量少的引物被逐渐耗尽,而超量的引物可继续直线扩增生成dna单链(gyllenstenanderlich,proc.natl.acad.sci.u.s.a.,1988,85:7652-7656)。该种方法具有较高的杂交灵敏度和特异性,且操作简便性更强,但其引物的比例需要优化,而且非特异扩增的机会增加,此外,dna单链分离过程需要依靠电泳,分离程序复杂,且电泳时常可见弥散条带,其耗时和不便是显而易见的。
上述几种分离方法均具有一定的局限性,因此,为了满足对dna单链分离的可操作性及经济性要求,现有技术中的dna单链采用的是集成化的提取工作站,将带有链亲和素的亲和连接体与dna双链结合,工作站具有抽滤针及配套的泵,结合后的dna亲和连接体通过抽滤吸附在抽滤针内滤膜下部,工作站配有轨道及相关系统,抽滤结束后将抽滤针移至盛有naoh的盘中,通过碱处理解双螺旋,得到dna单链,再次抽滤后清洗收集。抽滤针一般24根(4*6)为一组,使用时必须保证有足够量的样品或试剂以保证工作站的正常运行,因此这种dna单链的收集方式十分的不灵活,只能以固定量加入工作站进行工作,且大量的损失在多次的抽滤及转移过程中产生,这对微量收集十分不利,并且抽滤针组需要同时进行工作,这对工作站各部件都具有一定的体积要求,整个工作站占用空间很大。庞大的系统使得在dna单链分离操作过程中,微分离柱需要来回反复移液,操作十分繁琐,不仅分离周期长、效率低,且整体设备价格较贵,导致dna单链分离的费用高,还需要耗费大量的试剂及其他资源,极不经济。此外,该工作站中抽滤针为金属材质,价格昂贵,往往是处理后再重复使用,故易导致残留物之间的交叉污染,可靠性不高,对分离及检测结果的准确性均会造成一定的干扰和影响。且溶液抽取过程中会有部分残余溶液贴壁,使得一定量的目的dna单链不能被微分离柱吸附,导致获得的dna单链比例降低,影响了分离率,造成了浪费。因此,用于焦磷酸测序的高质量高效率的dna单链分离问题亟待解决。
技术实现要素:
针对现有技术存在的上述问题,本发明的目的是提供一种操作简便、快速检测的dna测序装置的控制系统及控制方法。
为实现上述发明目的,本发明采用的技术方案如下:
一种dna测序装置的控制系统,所述测序装置包括设有多个过滤柱的可旋转的分离盘;设有四个槽位的dntp试剂槽;固定设有多个加样针的加样架;用于支撑所述加样架移动的直线导轨;用于加样架转动的转轴;用于带动加样架上下移动的直线导轨;设有多个槽位的反应槽;用于采集每个槽位光信号的ccd相机;以及用于ccd相机转动的转盘;所述控制系统包括plc以及与plc连接并通过plc操作的光控制系统、位置控制系统、环境控制系统、流量控制系统和驱动控制系统;
所述光控制系统用于控制ccd相机的开或关;
所述位置控制系统包括多个位置传感器,通过位置传感器的信号控制加样架的转动角度、移动位置,以及ccd相机的转动角度;
所述环境控制系统包括位于反应槽内的温度传感器和加热器;用于检测溶液ph值的ph计;
所述流量控制系统包括总管道;与总管道一端连通且通向各部件的分管道;与总管道另一端连通的试剂管道;以及与总管道中段连通的溶液管道;所有管道都设有阀门;所述总管道上设有蠕动泵;
所述驱动控制系统包括用于驱动分离盘转动的电机;用于驱动转轴的电机;用于驱动ccd相机转盘的电机。
作为上述技术方案的进一步改进:
优选地,所述试剂管道包括datp管、dctp管、dgtp管、dttp管、dna聚合酶、荧光素管、atp管、aps管、缓冲液管和apyrase管,所述溶液管道为缓冲液管和水管。
优选地,所述总管道靠近分管道处设有除泡器。
优选地,所述试剂管道与一个第一多通换向阀的进口连接,所述第一多通换向阀具有十个进口和一个出口;所述总管道与分管道之间设有第二多通换向阀,所述第二多通换向阀具有一个进口和多个出口,所述缓冲液管和水管分别与第三多通换向阀的进口连接,所述第三多通换向阀具有两个进口和一个出口。
优选地,所述总管道靠近第一多通换向阀处设有第一蠕动泵,所述第三多通换向阀与总管道之间设有第二蠕动泵。
优选地,所述第一多通换向阀还具有一个备用的进口。
本发明的目的还在于提供了上述控制系统的控制方法,包括以下步骤:
s1、开启所述第一蠕动泵,依次开启所述第一多通换向阀的进口,开启所述第二多通换向阀中通向dntp试剂槽的分管道,将dntp试剂槽的四个槽位加入一定量的试剂;
s2、驱动所述分离盘旋转进行离心处理,过滤扩增、结合后的dna双链,得到待测dna单链;
s3、开启第二蠕动泵,开启所述第三多通换向阀的水管或缓冲液管进口,开启所述第二多通换向阀中通向过滤柱的分管道;用水或缓冲液清洗平衡ph值,至dna单链完全悬浮;
s4、开启所述第一蠕动泵,依次开启所述第一多通换向阀的进口,开启所述第二多通换向阀中通向反应槽的分管道,将荧光素和aps注入反应槽内;
s5、驱动加样架移动、转动,使加样针蘸取任意一种dntp试剂,并加入到测序反应液中,再使所述加样针41离开所述测序反应液;
s6、开启第二蠕动泵,开启所述第三多通换向阀的水管进口,清洗加样针;
s7、开启第一蠕动泵,根据设置依次开启所述第一多通换向阀的进口,开启所述第二多通换向阀中通向反应槽的分管道,依次加入dna聚合酶、atp硫酸化酶和核苷酸降解酶apyrase;
s8、4种dntp试剂任意或多个依待测序列而定以何种顺序加入测序反应液中,进行步骤s9或者重复步骤s5、s6和s7;
s9、控制ccd相机拍照,并通过plc呈现检测的谱图。
上述控制方法中,优选地,在所述步骤s7中,在抽取任意一种试剂后,开启所述第一蠕动泵,使所述第一多通换向阀的进口与盛放缓冲液的一个试剂瓶连通,用于抽取缓冲液形成位于相邻的两种试剂之间的液柱。
针对现有技术存在的上述问题,本发明dna测序装置的控制系统及控制方法的优势是:
(1)本方法简单易操作,获取目标样品时间短效率高,可用于痕量dna单链的收集与提取,样品损失几乎可以忽略,使用试剂少、对设备的要求低,有效简化了操作步骤,缩短了操作时间,降低了工作强度,提高了工作效率。
(2)本发明采用一结构简单的加样针即可实现dntp试剂的微量给样,摒弃了传动的中空针管抽喷式的给样方式,仅通过加样针在测序反应液内上下运动数次即可实现搅拌使得酶反应更为完全结果准确。
总之,本发明提供的dna测序装置的控制系统及控制方法减少了底物及酶系的用量,检测准确,反应速度快,集成化高,且可以同时检测一至多个snp位点以及单链dna片段,酶系及底物均打破了部分生产商的垄断,价格大大下降,分析装置结构精密,对待测dna片段及底物的用量要求低,降低了焦磷酸测序的检测成本,扩大了焦磷酸测序的使用范围。
附图说明
图1是本发明的dna测序装置的结构示意图。
图2是实施例中的用于焦磷酸的dna收集管的一使用优选实施例。
图3是实施例中过滤柱的结构示意图。
图4为实施例中加样架的加样针示意图。
图5为本发明的测序方法的液路结构示意图。
图中标号说明:
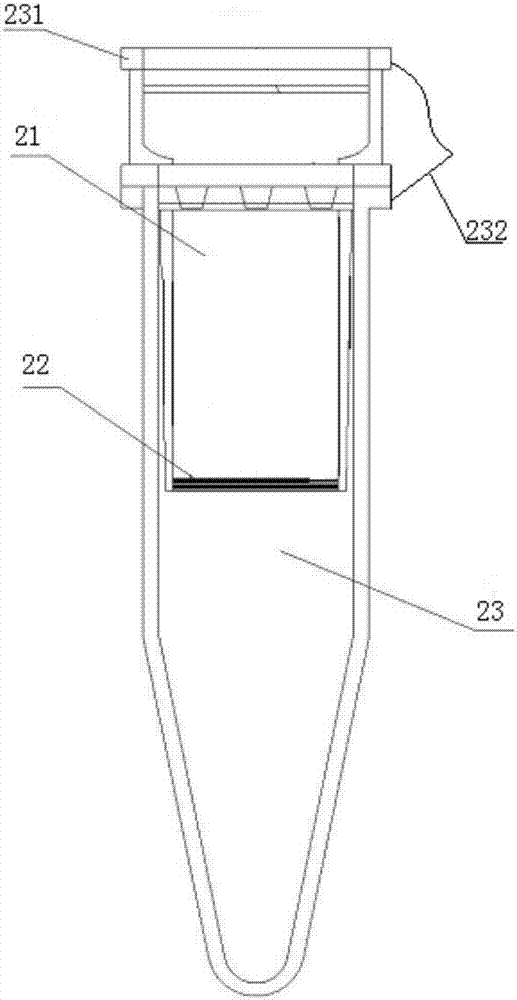
1、废液管道;2、分离盘;21、过滤柱;22、滤膜;23、收集管;231、上盖;232、连接带;3、反应槽;31、反应位;4、加样架;41、加样针;42、转轴;5、ccd相机;6、分析台;61、直线导轨;7、试剂槽;71、干燥槽;72、清洗槽;8、总管道;81、分管道;82、试剂管道;83、溶液管道;84、除泡器;85、第一多通换向阀;86、第二多通换向阀;87、第三多通换向阀;88、第一蠕动泵;89、第二蠕动泵。
具体实施方式
下面结合实施例对本发明作进一步详细、完整地说明。
实施例1
结合图1至图4详细说明本发明实施例的控制系统及控制方法的dna测序装置的结构。
本发明的dna测序装置是基于焦磷酸测序,可用于焦磷酸测序检测分析dna序列,待测序的dna序列为靶序列,将靶dna序列进行扩增后进行焦磷酸测序。
本实施例的基于焦磷酸测序的dna测序系统包括dna测序装置和控制区,dna测序装置包括样品区、反应区和检测区。样品区、反应区和检测区安装在分析台6上,样品区、反应区和检测区通过控制区监测、控制。整个分析过程采用机械手操作。
进行焦磷酸测序之前,需要先对带测序的dna模板进行扩增,以达到扩增要求的dna浓度。扩增引物设计时,扩增引物上带有亲和连接体,亲和连接体优选为生物素,亲和连接体优选地标记在靶dna的一端引物上,pcr扩增采用现有技术进行即可。
样品区包括分离盘2。分离盘2位于反应区的上方,在靶dna扩增后,靶dna为双链dna,焦磷酸测序需要单链dna,分离盘2包括至少一个dna分离区,dna分离区采用物理过滤的方式,分离区包括内部设有滤膜22的中空的过滤柱21,滤膜22为高分子纳米微球结构,由于高分子纳米微球间的孔径小于亲和连接体的直径,将带有亲和连接体标记的dna单链留在膜上,根据测序需要收集带标记的dna单链或其互补链,用于焦磷酸测序。
本实施例中,分离盘2设有多个收集管23,过滤柱21安装于收集管23中,滤膜22位于过滤柱21的一端的端部。过滤柱21下段的外径与收集管23的内径相同。
双链dna在碱解后将待亲和连接体的一条链留在滤膜22上,收集该条链只需在过滤柱21内加入收集液后收集;如需收集互补链,可将滤液收集后对滤液中的互补链进行收集。
如图3所示,收集管23用于收集离心时产生的废液,每个步骤后需要及时倾倒以防止交叉污染,收集管23的上端管身为圆柱体、下端为圆锥体,底部为球面状。作为另一种优选的实施方式,收集管23配置上盖231,上盖231通过连接带232可拆卸地连接在收集管23上,由于连接带232的连接,装入过滤柱21后上盖231也可紧密扣合在过滤柱21或收集管23上,上盖231的作用是在液体离心时防止液体飞溅出过滤柱21造成损失及污染。
本实施例中所优选的滤膜22材料为聚乙烯微球,其微球间孔隙优选为10μm,小于亲和连接体的直径,可直接通过物理过滤将带有亲和连接体的单链留在膜上,滤去未连接亲和连接体的一条链,其吸附洗脱效果好,dna回收率高,原料价格低廉环保。
为了本实施例中的滤膜22在吹打或离心过程中不发生移动,本实施例中还优选的在滤膜22上方设有压膜装置,压膜装置包括垫片和/或压膜架。待分离纯化的液体先通过垫片后与滤膜22接触,垫片优选为纤维材料,可耐受酸碱和大部分有机溶剂,对大部分的生物分子不会产生吸附。
优选地在垫片的上方,不与滤膜22接触的一侧,还设有压膜架,压膜架与过滤柱21体材料相同,通过机械压力压紧滤膜22,使滤膜22不会在吹打或离心等使用过程中发生移动,造成收集损失。
反应区包括加样部和反应槽3,加样部依次包括dntp试剂槽7、干燥槽71、清洗槽72、加样架4和安装于加样架4上的多个加样针41。分离盘2位于试剂槽7的侧上方。加样架4为圆盘形,加样架4通过直线导轨61可沿试剂槽7和反应槽3之间做往复运动。干燥槽71为真空干燥区。
试剂槽7设有四个槽位,用于焦磷酸测序的dntp包括datp、dctp、dgtp、dttp四种核酸底物,底物用于与靶dna杂交,并且需要在反应体系中加入相关酶系催化反应进行,具体为dna聚合酶,进行互补链合成反应,在互补链合成时得到的副产物焦磷酸转化成atp,在萤光素酶存在下使atp和萤光素反应,进行发光。如果发生互补链合成,则产生焦磷酸,结果发光,因此通过对其进行监视,可以得知产生互补链合成,即组入的碱基种类,由此确定dna序列。
加样架4通过转轴42做旋转运动,转轴42设有驱动电机。转轴42安装于直线导轨61上,转轴42带动加样架4做往复运动,加样架4通过另一组直线导轨在转轴42上做升降运动。加样针41为实心针,用于转移dntp试剂,加样针41固定于加样架4的通孔中。将dna单链按照反应体系的需要量转移至反应槽3中,加入酶系后依次加入dntp,加入顺序不限,对于每个位点,四种底物各加入一次。
检测区包括ccd相机5,ccd相机5位于反应槽3的中间。反应后如合成互补链,则发光,ccd相机5用于检测判断该位点是否进行反应并发光,进而判断出该位点的碱基序列。
加样架4位于反应槽3上;反应槽3上设有多个反应位31,反应位31中有测序反应液。反应位31沿反应槽3的圆心呈一系列同心圆均匀排列,反应位31由透明材质构成,反应位31延伸入ccd相机5。
如图4所示,加样针41的端面为半圆体;采用上述的加样装置检测已知序列的样品(序列为caatattcgccaggt),其中加样针41直径为1.5mm,表面光洁度为ra0.8;加样方法包括:步骤a,通过加样针41插入反应槽3的dntp试剂使得加样针41外周附着所述dntp试剂;步骤b,将附着所述dntp试剂的加样针41插入反应位31中,再使加样针41离开测序反应液;所述步骤a中所述加样针41离开所述dntp试剂液时的移动速度为10cm/s,所述步骤b中所述加样针41离开所述测序反应液时的移动速度为1cm/s,在测序反应液内上下运动8次后离开测序反应液。
本发明的基于焦磷酸测序的dna测序系统的设置方式可以更好地传递底物与dna,减少传递过程中的损耗。
反应区的形状不限,分析装置内的具体结构可以根据检测的需要进行设计调整。
底物加样至反应区中,此时反应区中已加入待测dna单链及其他所需反应体系内试剂,以检测所需5ul的量计算,反应体系如下:
酶混合物包括:
底物混合物包括:
aps0.4mg/l;
萤火虫荧光素0.4mmol/l。
反应过程中,在每一步选择对酶活性而言最适宜的ph值进行反应,反应后需要对反应体系内的ph值进行调整以适应其他反应,具体反应体系中的反应条件本领域普通技术人员可根据现有技术得出。例如,当洗涤步骤中包含腺苷三磷酸双磷酸酶以去除过量核苷酸种类和atp时,因为处理步骤的连续性,缓冲液可与腺苷三磷酸双磷酸酶一起使用,其ph能使腺苷三磷酸双磷酸酶洁性水平最佳化。然后,在下一个核苷酸掺入步骤中使用聚合酶,可使用具有对聚合酶而言的最佳ph条件的不同缓冲液,以便使聚合酶洁性最佳化。另外,每种最佳缓冲液可包括对于每种酶而言的优选抗衡离子,例如对于腺苷三磷酸双磷酸酶缓冲液而言的ca2+和对于聚合酶缓冲液而言的mg2+。
反应体系中,检测序列为caatattcgccaggt,其中依次加入dntp的顺序为tcatattcgccagt,其中首次加入t时未出峰,以及后续与实际序列不符的dntp试剂也未出峰,其测得的序列为caatattcgccaggt,与样品实际序列完全一致,精确度为100%;检测单个碱基的时间小于1.5分钟,此序列大约耗时25分钟,且最小加样量可达0.1μl。
测序结果通过生物发光来进行判断,其他可以检测生物发光的装置均可使用为检测装置,在此不做限制。
以下结合图1至图5,详细说明上述dna测序装置的控制系统及控制方法。
本发明的dna测序装置的控制系统,包括plc以及与plc连接并通过plc操作的光控制系统、位置控制系统、环境控制系统、流量控制系统和驱动控制系统。
光控制系统用于控制ccd相机5的开或关。位置控制系统包括多个位置传感器,通过位置传感器的信号控制加样架4的转动角度、移动位置,以及ccd相机5的转动角度。环境控制系统包括位于反应槽3内的温度传感器和加热器以及用于检测溶液ph值的ph计。驱动控制系统包括用于驱动分离盘转动的电机;用于驱动转轴的电机;用于驱动ccd相机转盘的电机。
流量控制系统包括总管道8;与总管道8一端连通且通向各部件的分管道81;与总管道8另一端连通的试剂管道82;以及与总管道8中段连通的溶液管道83;所有管道都设有阀门;总管道8上设有蠕动泵;总管道8靠近分管道81处设有除泡器84。
试剂管道82包括datp管、dctp管、dgtp管、dttp管、dna聚合酶、荧光素管、atp管、aps管、缓冲液管和apyrase管,溶液管道83为缓冲液管和水管,缓冲液用于稀释试剂和冲洗零部件。试剂管道82与一个第一多通换向阀85的进口连接,第一多通换向阀85具有十一个进口和一个出口,多出来的一个进口备用。第一多通换向阀85的出口通过第一蠕动泵88与总管道8连接。总管道8与分管道81之间设有第二多通换向阀86,第二多通换向阀86具有一个进口和多个出口。缓冲液管和水管分别与第三多通换向阀87的进口连接,第三多通换向阀87具有两个进口和一个出口。第三多通换向阀87的出口通过第二蠕动泵89与总管道8连接。总管道8在靠近分管道81处设有废液管道1,反应槽3的底部设有出液口与废液管道1连接。整体液路依靠第一蠕动泵88和第二蠕动泵89提供液流压力,第一蠕动泵88和第二蠕动泵89分别由相应的步进电机驱动。除泡器84用于消减溶解的气体,精确控制进入反应槽3的试剂和溶液的量。
本实施例中,上述控制系统的控制方法,包括以下步骤:
s1、开启第一蠕动泵88,依次开启第一多通换向阀85的进口,开启第二多通换向阀86中通向dntp试剂槽的分管道81,将dntp试剂槽的四个槽位加入一定量的试剂。加入试剂时,每加完一种试剂后,需清洗管道,避免相邻试剂之间的干扰,提高测序的精确度。
s2、驱动分离盘2旋转进行离心处理,除去多余的溶液,洗去残留的扩增试剂,过滤扩增、结合后的dna双链;再想过滤柱41中加入naoh与nacl解开双链螺旋。
s3、开启第二蠕动泵89,开启第三多通换向阀87的水管或缓冲液管进口,开启第二多通换向阀86中通向过滤柱的分管道81;用水或缓冲液洗去残留的naoh,平衡ph值至中性,再加入水,至dna单链完全悬浮后吸出用于焦磷酸测序或4℃密封保存。
s4、开启第一蠕动泵88,依次开启第一多通换向阀85的进口,开启第二多通换向阀86中通向反应槽的分管道81,将荧光素和aps注入反应槽3内作为反应底物,再将步骤s3中得到的待测单链放入反应槽3内。
s5、驱动加样架4移动,使加样针蘸取任意一种dntp试剂使得加样针外周附着dntp试剂;驱动加样架4移动,将附着dntp试剂的加样针插入测序反应液中,再使加样针41离开测序反应液;反应过程中通过温度传感器和加热器控制反应槽3的温度,保持酶的活性。
s6、开启第二蠕动泵89,开启第三多通换向阀87的水管进口,清洗加样针。
s7、开启第一蠕动泵88,根据设置依次开启第一多通换向阀85的进口,开启第二多通换向阀86中通向反应槽的分管道81,依次加入dna聚合酶、atp硫酸化酶和核苷酸降解酶apyrase;在抽取任意一种试剂后,开启第一蠕动泵88,使第一多通换向阀85的进口与盛放缓冲液的一个试剂瓶连通,用于抽取缓冲液形成位于相邻的两种试剂之间的液柱。
s8、4种dntp试剂任意或多个依待测序列而定以何种顺序加入测序反应液中,进行步骤s9或者重复步骤s5、s6和s7。
本实施例中,仅检测单个碱基是否变异,可根据已知的前后序列先确定待测碱基位置后,依次重复上述步骤4次加样(加入4种dntp试剂),分别加入同一测序反应液中检测该碱基。
s9、驱动ccd相机转动角度,使ccd相机对准正在进行反应的反应槽3,控制ccd相机拍照,并通过plc呈现检测的谱图。
s10、测序后,开启反应槽3的出液口,将废液排入废液管道1.
本发明所述实施方案的控制系统和方法中,使用计算机可读介质进行一些设计、分析或其它操作,这样的介质存储有用于在计算机系统上执行的指令。
在相同或替代实施方案中,在计算机上的应用可使用包括所谓“命令行界面”(通常称为cli)在内的界面。cli通常提供基于应用与用户间相互作用的文本。通常,命令行界面通过显示器以文本行形式显示输出并接收输入。例如,某些执行过程可包括所谓的"命令行解释程序(shell)"例如相关领域普通技术人员己知的unix命令行解释程序(unixshell),或使用面向对象型程序体系结构的microsoftwindowspowershell,例如microsoft.net框架。
相关领域普通技术人员将会知道,界面可包括一种或多种gui、cli或其组合。
处理器通常执行操作系统,操作系统可选择现有技术中的任何操作系统,只要本领域技术人员可用于处理检测结果及数据等工作均认为可以纳入保护范围之内。
本实施例中的dna分离过程中所涉及的溶液、参数及其他分离条件,均为本实施例中的优选实施方案,任何参照现有技术可以起到相应作用的实验条件、参数及溶液均可用于本发明中所涉及的分离纯化过程,本实施例中的具体参数及溶液选择不应作为对本发明的限制。
最后有必要在此说明的是:以上实施例只用于对本发明的技术方案作进一步详细地说明,不能理解为对本发明保护范围的限制,本领域的技术人员根据本发明的上述内容作出的一些非本质的改进和调整均属于本发明的保护范围。最后有必要在此说明的是:以上实施例只用于对本发明的技术方案作进一步详细地说明,不能理解为对本发明保护范围的限制,本领域的技术人员根据本发明的上述内容作出的一些非本质的改进和调整均属于本发明的保护范围。
- 还没有人留言评论。精彩留言会获得点赞!