一种呱西替柳的制备方法与流程
本发明属于药物合成
技术领域:
,涉及一种呱西替柳的合成方法和精制方法。
背景技术:
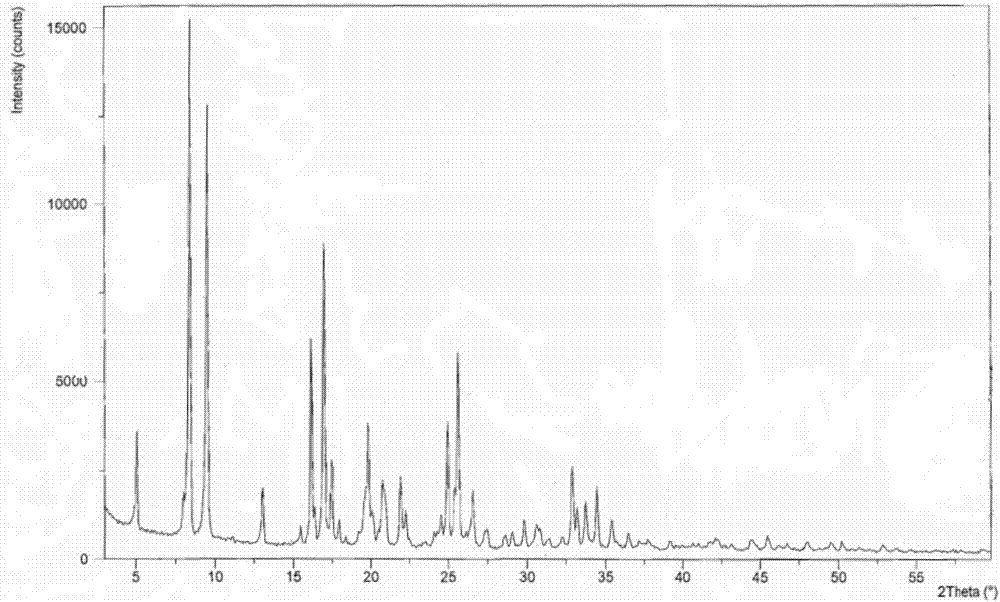
:呱西替柳是水杨酸类衍生物,结构式如下。呱西替柳由德国拜耳公司于1979年研发成功,1982年在意大利上市,是一种非甾体类解热镇痛药,兼具消炎、镇咳和祛痰作用,临床上用于治疗伴有发热、疼痛的急慢性呼吸道炎症及粘膜炎症等。制备呱西替柳的方法文献报道主要有三种。方法一,以乙酰水杨酸为原料,制备乙酰水杨酰氯,同愈创木酚钠进行酯化得到呱西替柳,见合成路线1(cn1337391;呱西替柳的合成工艺改进,张大永,华西药学杂志,1998,1(3):182-183)合成路线1此路线合成步骤长,所用起始原料乙酰水杨酸(阿司匹林)价格比水杨酸高。反应用到氯化亚砜或者光气为酰氯化试剂。光气为有毒气体,损害呼吸道,可导致化学性支气管炎、肺炎、肺水肿。氯化亚砜具有强挥发性,对人体呼吸道刺激也比较强烈。反应形成乙酰水杨酰氯,必须引入惰性气体避水保存,在工业中对设备密封要求高。此外乙酰水杨酰氯与愈创木酚钠反应为两相反应,需要加入相转移催化剂促进反应进行以提高收率。方法二,以水杨酸和愈创木酚为起始原料,进行酯化反应得到水杨酸愈创木酚酯,然后对羟基进行乙酰化得到呱西替柳,见合成路线2(呱西替柳的合成,孙长安,中国医药工业杂质,1990,21(9);呱西替柳合成方法的改进,赵书清,黑龙江大学自然科学学报,1993年3月,第10卷第1期;cn104744261)合成路线2该路线水杨酸与愈创木酚酯化反应需要的温度高,反应时间长,能耗大,导致生产成本高。且酯化反应用三氯氧磷为酯化剂,刺激性较强,后处理需要外循环冷水降低体系温度,否则放热大导致温度过高,易发生危险。方法三,以乙酰水杨酸为原料,经cdi活化后与愈创木酚反应得到呱西替柳,见合成路线3(journalofchemicalandpharmaceuticalresearch,2012,4(1):580-582)合成路线3该路线简短,反应条件温和。但起始原料阿司匹林价格较水杨酸高,且反应后处理需要浓缩高沸点溶剂dmf,需要的温度和真空度高,能耗大。技术实现要素:本发明采用价格低廉的水杨酸为起始原料,经n,n’-羰基二咪唑活化后与愈创木酚反应得到水杨酸愈创木酚酯,再进行羟基乙酰化反应得到呱西替柳。本发明在已有合成路线的基础上对合成方法进行了改进,避免使用三氯氧磷、氯化亚砜、光气等毒性较大、强刺激性的试剂,同时无需使用相转移催化剂,使反应条件温和,操作更加简单,产品纯度更高。经过实验意外发现采用本发明的制备方法得到了呱西替柳的一种新晶型,相对已有晶型提高了呱西替柳水溶性及其稳定性,有关物质含量显著降低,大大提高了用药的安全性。为了达到上述目的,采用的技术方案为:本发明提供一种呱西替柳的制备方法,包括如下步骤:s1:酯化反应:以水杨酸为起始原料,经n,n’-羰基二咪唑(cdi)活化后与愈创木酚反应得到中间体水杨酸愈创木酚酯;s2:羟基乙酰化反应:中间体水杨酸愈创木酚酯与乙酰氯反应,发生羟基乙酰化反应,得到产物呱西替柳粗品;反应式如下:s3:将呱西替柳粗品溶于乙醇,加热溶解后,加入不良溶剂析出固体,继续降温析晶得到呱西替柳晶型;包括如下步骤:1)将呱西替柳粗品溶于乙醇,加热至60~70℃溶解,得到溶液a;2)加入活性炭,搅拌30min,过滤得到溶液b;3)60℃下滴加不良溶剂至析出固体,继续降温至10℃析晶1小时;4)过滤,乙醇淋洗得到白色固体;5)将步骤4)所得固体置于真空干燥箱,抽真空,干燥,得到呱西替柳晶型。优选地,s1中,水杨酸:cdi:愈创木酚的摩尔比为1:1.05~1.2:1.0~1.2;反应温度为10~35℃。优选地,s1所用反应溶剂为thf、dmf或dcm中的一种。优选地,s2中水杨酸愈创木酚酯在缚酸剂作用下与乙酰氯反应,反应温度为0~30℃;水杨酸愈创木酚酯:乙酰氯:缚酸剂的摩尔比为1:1.0~1.2:1.1~1.5。优选地,s2所用的缚酸剂选自三乙胺、二异丙基乙胺、二乙胺、二异丙基胺中的一种或几种。优选地,s2中所用的反应溶剂为dcm、thf或dmf中的一种。优选地,s3中步骤1)中呱西替柳粗品:乙醇质量体积比为1:1.5-2.5(g/ml);步骤5)所述的干燥在30-50℃条件下进行。优选地,s3中步骤3)滴加的不良溶剂为正己烷。本发明的制备方法是在已有合成路线的基础上对合成方法进行了改进,避免使用三氯氧磷、氯化亚砜、光气等毒性较大、强刺激性的试剂,同时无需使用相转移催化剂,使反应条件温和,操作更加简单,产品纯度更高。本发明中,所述的呱西替柳粗品可以为待进一步纯化的呱西替柳固体混合物。呱西替柳粗品也可以为市售原料或通过现有技术方法制备得到,所得的晶型结果均在误差范围内,均为新晶型b。一个原料药的不同晶型可以有不同的化学和物理特性,包括熔点、化学反应性、表观溶解度、溶解速率、光学和机械性质、蒸气压和密度。这些特性可以直接影响原料药和制剂的处理和/或生产,并且会影响制剂的稳定性、溶解度和生物利用度。当化合物存在多晶型时,由于特定多晶型物具有特异性的热力学性质和稳定性,因此在制备的过程中,了解在各个剂型中应用的化合物的晶型是重要的,以保证生产过程应用相同形态的药物活性化合物。因此,保持药物活性化合物是单一的晶型或是一些晶型的已知混合物是必要的。现有技术中已有大量的文献及专利报道了呱西替柳的合成方法,大多数采用乙醇重结晶或精制,得到晶体:如陈小全,仇玉芹等.超声波作用下合成药物呱西替柳.医药农药.2009年第02期;陈立钦,白崇荣,高锁.呱西替柳的催化合成.武汉化工学院学报,2001年第23卷第4期;孙长安,黄建军.呱西替柳的合成.中国医药工业杂志.1990年21卷第9期;张大永,徐鸣夏.呱西替柳的合成工艺改进.华西药学杂志,1998年13卷第3期;王秋芬,郑更修等.呱西替柳的合成工艺改进;赵书清,邢有权等.呱西替柳合成方法的改进.黑龙江大学自然科学学报.1993年第10卷第1期;祁铁流,薛荣,韦玮.呱西替柳合成方法的改进.海军医高专学报,1997年第19卷第3期;cn104744261a,一种呱西替柳的生产方法,张云。也有使用chcl3析出晶体:如《醋柳愈酯的相转移催化合成》天津化工,2001年第1期,何明华,林家逊。cn103102271b公开了一种呱西替柳的工业化制备方法,其创新在于向反应液中滴加溶剂甲醇、乙醇、异丙醇、正丙醇或异丁醇,冷却直接析出产品,是将常规处理中把反应液倒入冰水中和再次用乙醇重结晶两步合并一步,简化操作,提高了安全性。晶体的形成机理很复杂,一个新晶体的获得也具有很大的偶然性,有时不同的溶剂、在不同的结晶条件下会产生相同的晶体结构。某些特定晶型也并非一定会获得更加有利的理化性质。药物的稳定性、吸湿性、溶解性、生物活性、毒性等性质会因晶型的不同而产生巨大的差异。研究表明,在x—射线粉末衍射图谱中,由新晶型得到的衍射谱图对于特定的晶型往往是特征性的,其中谱带(尤其是在低角度)的相对强度可能会因为结晶条件、粒径和其它测定条件的差异而产生的优势取向效果而变化。因此,衍射峰的相对强度对所针对的晶型并非是特征性的,判断是否与已知的晶型相同时,更应该注意的是峰的相对位置而不是它们的相对强度。本申请进行了大量的试验,发现采用现有技术的方法用乙醇或chcl3重结晶或采用cn1337391a方法用石油醚-乙酸乙酯重结晶或采用cn103102271b的方法用甲醇、乙醇、异丙醇、正丙醇或异丁醇冷却析晶,或将甲醇、乙醇、异丙醇、正丙醇、异丁醇及chcl3两种、三种或多种溶剂混合结晶或降温析晶或采用现有技术的工艺制备晶体,均得到晶型a化合物,且晶型a化合物均存在水溶性不好,总杂及单杂含量高。本发明经过大量的试验研究,惊喜地发现采用本发明的制备方法得到了一种与现有技术晶型a具有明显不同峰的相对位置的新的呱西替柳晶型b化合物。本发明的呱西替柳新晶型b化合物相对已有晶型提高了呱西替柳水溶性及其稳定性,有关物质含量显著降低,大大提高了用药的安全性。下面通过对本发明提供的呱西替柳晶型b化合物进行研究来解释和说明本发明技术方案:1、晶型检测取本发明制备得到的呱西替柳结晶b,使用cu-kα射线测量得到的x-射线粉末衍射图如图1所示,其以2θ±0.2衍射角表示的x-射线粉末衍射图在5.0486°、8.3928°、9.4999°、11.0938°、13.0428°、15.4407°、16.1385°、16.9587°、17.4526°、17.9250°、19.8174°、20.1244°、20.7524°、21.9069°、22.2502°、23.5129°、24.0635°、24.9467°、25.6333°、26.5561°、27.4492°、28.5748°、29.0483°、29.8173°、30.5616°、30.8142°、31.3825°、32.2431°、32.8643°、33.2108°、33.7183°、34.4484°、35.3625°、36.4592°、37.7823°、39.1404°、42.1802°、43.1244°、44.4461°、45.4795°、46.7445°、47.9937°、49.5318°、50.1831°、52.8140°、53.7477°处显示有特征峰。2、差热分析及热重分析对本发明制备的呱西替柳晶体进行差热和热重分析,结果如附图2所示;结果表明,本品在100℃处有吸热峰。本品经熔点测定:99~102℃,从侧面证明了其为一种不同的晶型。3、水分分析采用卡式水分测定仪测定,本发明的呱西替柳结晶的含水量为0.02%。与现有技术相比,本发明具有如下优点:(1)本发明的合成方法避免了使用三氯氧磷、氯化亚砜、光气等毒性较大、强刺激性的试剂,且大大降低了反应温度,使反应条件温和,操作简单。(2)本发明的制备方法得到了呱西替柳的一种新晶型b,相对已有晶型提高了呱西替柳水溶性及其稳定性,有关物质含量显著降低,大大提高了用药的安全性。附图说明图1为本发明制备的呱西替柳晶型b化合物的射线粉末衍射图谱;图2本发明制备的呱西替柳晶型b化合物的tg-dsc图谱。图3cn1337391a方法制备的呱西替柳hplc检测图谱。图4journalofchemicalandpharmaceuticalresearch,2012,4(1):580-582方法制备的呱西替柳hplc检测图谱。图5cn103102271a方法制备的呱西替柳hplc检测图谱。图6吉林金恒制药股份有限公司精制品,批号140225056呱西替柳hplc检测图谱。图7本发明实施例1制备的呱西替柳hplc检测图谱。图8本发明实施例2制备的呱西替柳hplc检测图谱。具体实施例以下用实施例对本发明的技术方案进行详细说明,将有助于对本发明的技术方案的优点、效果有更进一步的了解,实施例不限定本发明的保护范围,本发明的保护范围由权利要求来决定。实施例1第一步:中间体水杨酸愈创木酚酯的制备将水杨酸(138g,1.0mol)和愈创木酚(136.6g,1.1mol)加入到1000mlthf中,搅拌溶解后分批加入cdi(194.6g,1.2mol)。加完室温下反应3小时。浓缩除去溶剂后加入600ml乙酸乙酯,分别以饱和碳酸氢钠溶液(2×400ml)和水(400ml)洗涤。浓缩去溶剂,加乙醇400ml浓缩至350ml,冷却析晶,过滤,滤饼于45℃下真空干燥得到中间体水杨酸愈创木酚酯(171g,收率70%,hplc纯度大于98%)。第二步:呱西替柳的制备将中间体水杨酸愈创木酚酯(97.7g,400mmol)溶于350ml干燥的dcm中,加入tea(60.7g,600mmol),冰浴降温30min后滴加乙酰氯(33.0g,420mmol)。滴完室温下搅拌反应,tlc检测反应完成后,反应液分别以5%hcl溶液(2×200ml)、纯化水(200ml)和食盐水(150ml)洗涤。浓缩去溶剂,加乙醇200ml浓缩至170ml,冷却析晶,过滤,45℃真空干燥得到呱西替柳(100.8g,收率88%,hplc纯度大于99%)。第三步:呱西替柳的精制将上步所得呱西替柳50g溶于100ml乙醇,加热至60-70℃溶解,加入0.5g活性炭脱色后过滤,滤液于60℃下滴加正己烷至固体析出,继续降温至10℃析晶1小时后过滤,乙醇淋洗,45℃真空干燥得到47.2g呱西替柳,hplc纯度99.8%。实施例2第一步:中间体水杨酸愈创木酚酯的制备将水杨酸(138g,1.0mol)和愈创木酚(124.1g,1.0mol)加入到1000mldmf中,搅拌溶解后分批加入cdi(170.3g,1.05mol)。加完室温下反应3小时。浓缩除去溶剂后加入600ml乙酸乙酯,分别以饱和碳酸氢钠溶液(2×400ml)和水(400ml)洗涤。浓缩去溶剂,加乙醇400ml浓缩至350ml,冷却析晶,过滤,滤饼于45℃下真空干燥得到中间体水杨酸愈创木酚酯(158.8g,收率65%,hplc纯度大于98%)。第二步:呱西替柳的制备将中间体水杨酸愈创木酚酯(97.7g,400mmol)溶于350ml干燥的thf中,加入dipea(56.9g,440mmol),冰浴降温30min后滴加乙酰氯(34.5g,439mmol)。滴完室温下搅拌反应,tlc检测反应完成后,反应液分别以5%hcl溶液(2×200ml)、纯化水(200ml)和食盐水(150ml)洗涤。浓缩去溶剂,加乙醇200ml浓缩至170ml,冷却析晶,过滤,45℃真空干燥得到呱西替柳(98.6g,收率86%,hplc纯度大于99%)。第三步:呱西替柳的精制将上步所得呱西替柳50g溶于75ml乙醇,加热至60-70℃溶解,加入0.5g活性炭脱色后过滤,滤液于60℃下滴加正己烷至固体析出,继续降温至10℃析晶1小时后过滤,乙醇淋洗,50℃真空干燥得到46.9g呱西替柳,hplc纯度99.8%。实施例3第一步:中间体水杨酸愈创木酚酯的制备将水杨酸(138g,1.0mol)和愈创木酚(149g,1.2mol)加入到1000mldcm中,搅拌溶解后分批加入cdi(178.4g,1.1mol)。加完室温下反应3小时。浓缩除去溶剂后加入600ml乙酸乙酯,分别以饱和碳酸氢钠溶液(2×400ml)和水(400ml)洗涤。浓缩去溶剂,加乙醇400ml浓缩至350ml,冷却析晶,过滤,滤饼于45℃下真空干燥得到中间体水杨酸愈创木酚酯(153.8g,收率63%,hplc纯度大于98%)。第二步:呱西替柳的制备将中间体水杨酸愈创木酚酯(97.7g,400mmol)溶于350ml干燥的dmf中,加入二乙胺(35.1g,480mmol),冰浴降温30min后滴加乙酰氯(33.0g,400mmol)。滴完室温下搅拌反应,tlc检测反应完成后,反应液分别以5%hcl溶液(2×200ml)、纯化水(200ml)和食盐水(150ml)洗涤。浓缩去溶剂,加乙醇200ml浓缩至170ml,冷却析晶,过滤,45℃真空干燥得到呱西替柳(95.1g,收率83%,hplc纯度大于98%)。第三步:呱西替柳的精制将上步所得呱西替柳50g溶于90ml乙醇,加热至60-70℃溶解,加入0.5g活性炭脱色后过滤,滤液于60℃下滴加正己烷至固体析出,继续降温至10℃析晶1小时后过滤,乙醇淋洗,30℃真空干燥得到46.3g呱西替柳,hplc纯度99.5%。实施例4第一步:中间体水杨酸愈创木酚酯的制备将水杨酸(138g,1.0mol)和愈创木酚(130.4g,1.05mol)加入到1000mlthf中,搅拌溶解后分批加入cdi(186.5g,1.15mol)。加完室温下反应3小时。浓缩除去溶剂后加入600ml乙酸乙酯,分别以饱和碳酸氢钠溶液(2×400ml)和水(400ml)洗涤。浓缩去溶剂,加乙醇400ml浓缩至350ml,冷却析晶,过滤,滤饼于45℃下真空干燥得到中间体水杨酸愈创木酚酯(183g,收率75%,hplc纯度大于98%)。第二步:呱西替柳的制备将中间体水杨酸愈创木酚酯(97.7g,400mmol)溶于350ml干燥的thf中,加入二异丙基胺(56.7g,560mmol),冰浴降温30min后滴加乙酰氯(37.7g,480mmol)。滴完室温下搅拌反应,tlc检测反应完成后,反应液分别以5%hcl溶液(2×200ml)、纯化水(200ml)和食盐水(150ml)洗涤。浓缩去溶剂,加乙醇200ml浓缩至170ml,冷却析晶,过滤,45℃真空干燥得到呱西替柳(97.3g,收率85%,hplc纯度大于99%)。第三步:呱西替柳的精制将上步所得呱西替柳50g溶于110ml乙醇,加热至60-70℃溶解,加入0.5g活性炭脱色后过滤,滤液于60℃下滴加正己烷至固体析出,继续降温至10℃析晶1小时后过滤,乙醇淋洗,40℃真空干燥得到48.5g呱西替柳,hplc纯度99.8%。实施例5第一步:中间体水杨酸愈创木酚酯的制备将水杨酸(138g,1.0mol)和愈创木酚(142.8g,1.15mol)加入到1000mldcm中,搅拌溶解后分批加入cdi(194.6g,1.2mol)。加完室温下反应3小时。浓缩除去溶剂后加入600ml乙酸乙酯,分别以饱和碳酸氢钠溶液(2×400ml)和水(400ml)洗涤。浓缩去溶剂,加乙醇400ml浓缩至350ml,冷却析晶,过滤,滤饼于45℃下真空干燥得到中间体水杨酸愈创木酚酯(166g,收率68%,hplc纯度大于98%)。第二步:呱西替柳的制备将中间体水杨酸愈创木酚酯(97.7g,400mmol)溶于350ml干燥的dmf中,加入tea(52.6g,520mmol),冰浴降温30min后滴加乙酰氯(36.1g,460mmol)。滴完室温下搅拌反应,tlc检测反应完成后,反应液分别以5%hcl溶液(2×200ml)、纯化水(200ml)和食盐水(150ml)洗涤。浓缩去溶剂,加乙醇200ml浓缩至170ml,冷却析晶,过滤,45℃真空干燥得到呱西替柳(103.1g,收率90%,hplc纯度大于99%)。第三步:呱西替柳的精制将上步所得呱西替柳50g溶于125ml乙醇,加热至60-70℃溶解,加入0.5g活性炭脱色后过滤,滤液于60℃下滴加正己烷至固体析出,继续降温至10℃析晶1小时后过滤,乙醇淋洗,45℃真空干燥得到47.0g呱西替柳,hplc纯度99.8%。实施例6第一步:中间体水杨酸愈创木酚酯的制备将水杨酸(138g,1.0mol)和愈创木酚(136.6g,1.1mol)加入到1000mlthf中,搅拌溶解后分批加入cdi(194.6g,1.2mol)。加完室温下反应3小时。浓缩除去溶剂后加入600ml乙酸乙酯,分别以饱和碳酸氢钠溶液(2×400ml)和水(400ml)洗涤。浓缩去溶剂,加乙醇400ml浓缩至350ml,冷却析晶,过滤,滤饼于45℃下真空干燥得到中间体水杨酸愈创木酚酯(171g,收率70%,hplc纯度大于98%)。第二步:呱西替柳的制备将中间体水杨酸愈创木酚酯(97.7g,400mmol)溶于350ml干燥的dcm中,加入tea(60.7g,600mmol),冰浴降温30min后滴加乙酰氯(33.0g,420mmol)。滴完室温下搅拌反应,tlc检测反应完成后,反应液分别以5%hcl溶液(2×200ml)、纯化水(200ml)和食盐水(150ml)洗涤。浓缩去溶剂,加乙醇200ml浓缩至170ml,冷却析晶,过滤,45℃真空干燥得到呱西替柳(99.9g,收率87%,hplc纯度大于99%)。第三步:呱西替柳的精制将上步所得呱西替柳50g溶于100ml95%乙醇,加热至60℃溶解,加入0.5g活性炭脱色后过滤,滤液冷却析晶后继续降温至10℃养晶1小时后过滤,95%乙醇淋洗,45℃真空干燥得到46.5g呱西替柳,hplc纯度99.7%。下面通过实验例进一步说明本发明:对比1:cn1337391a将乙酰水杨酸溶于苯,加入催化剂dmf,再加入液体光气制备乙酰水杨酰氯的苯溶液;取愈创木酚溶于15%氢氧化钠溶液,再加入苯,然后将制备的乙酰水杨酰氯的苯溶液加到所述愈创木酚的苯-水混合液中。酯化完成后分出有机层,浓缩得到粗品,石油醚-乙酸乙酯重结晶得到纯品。对比2:呱西替柳的合成,孙长安,中国医药工业杂志,1990,21(9)水杨酸愈创木酚酯的制备同对比文件2。将水杨酸愈创木酚酯、醋酐、浓硫酸按摩尔比1:5:0.04的比例混合,60℃反应完成后搅拌下滴入冰盐水中,放置2-3小时,待析晶完全后过滤,水洗至中性,乙醇精制得到呱西替柳。对比3:journalofchemicalandpharmaceuticalresearch,2012,4(1):580-582以乙酰水杨酸为原料,经cdi活化后与愈创木酚反应得到呱西替柳粗品,95%乙醇精制得到纯品。对比4:cn103102271a催化剂dmap存在下,加入三氯氧磷使水杨酸和愈创木酚在60℃左右缩合,制得水杨酸愈创木酚酯后以醋酸酐乙酰化制得呱西替柳粗品,加入异丙醇淬灭反应并以异丙醇精制得到纯品。对比5:吉林金恒制药股份有限公司精制品,批号140225056。对比6:参照文献(陈小全,仇玉芹等.超声波作用下合成药物呱西替柳.医药农药.2009年第02期)制备的呱西替柳晶型a。对比7:参照文献(陈立钦,白崇荣,高锁.呱西替柳的催化合成.武汉化工学院学报.2001年第23卷第4期)制备的呱西替柳晶型a。对比8:参照文献(孙长安,黄建军.呱西替柳的合成.中国医药工业杂志.1990年第21卷第9期)制备的呱西替柳晶型a。对比9:参照文献(张大永,徐鸣夏.呱西替柳的合成工艺改进.华西药学杂志.1998年13卷第3期)制备的呱西替柳晶型a。对比10:参照文献(王秋芬,郑更修等.呱西替柳的合成工艺改进)制备的呱西替柳晶型a。对比11:参照文献(赵书清,邢有权等.呱西替柳合成方法的改进.黑龙江大学自然科学学报.1993年第10卷第1期)制备的呱西替柳晶型a。对比12:参照文献(祁铁流,薛荣,韦玮.呱西替柳合成方法的改进.海军医高专学报.1997年第19卷第3期)制备的呱西替柳晶型a。对比13:参照专利cn104744261a制备的呱西替柳晶型a。对比14:参照文献(何明华,林家逊.醋柳愈酯的相转移催化合成.天津化工.2001年第1期)制备的呱西替柳晶型a。对比15:参照专利103102271b制备的呱西替柳晶型a。实验例1:晶型化合物制备试验条件筛选采用本发明的精制方法操作,具体如下:将呱西替柳粗品溶于溶剂a,加热至溶解,加入活性炭脱色后过滤,滴加不良溶剂b至固体析出,降温至析晶完全,过滤,45℃真空干燥得到呱西替柳。表1-1试验条件筛选结果表1-2试验条件筛选结果表1-3试验条件筛选结果实验例2:溶解度测定参照中国药典2015年版二部凡例测定其溶解性,方法:取本品适量,分别加入水,每隔5分钟强力振摇30秒钟,观察30分钟内的溶解情况,即得,结果见表2。表2本发明的晶型和对照品在水中溶解性试验结果将上述实施例1-3溶解的水溶液样品在25℃恒温搅拌72小时,取样5ml。样品经0.45μm微孔滤膜过滤,弃去初滤液,取续滤液20μl测定药物含量即为水中溶解度(mg/ml)。结果见表3:表3本发明晶型与现有技术晶型在水中的溶解度对比从表2-3可以看出,25℃下,本发明呱西替柳晶型b化合物的在水中的溶解度与现有技术相比,有显著提高,取得了意料不到的技术效果。实验例3:有关物质检测检测方法:所用仪器:高效液相色谱仪、十八烷基硅烷键合硅胶柱、分析天平、烧杯、容量瓶。流动相:甲醇-水(75:50);检测波长:240nm。供试品溶液:取本品约10mg,精密称定,置25ml容量瓶中,加流动相溶解并稀释至刻度,摇匀,滤过,取续滤液作为供试品溶液。对照溶液:精密量取供试品溶液适量,加流动相制成每1ml中含4μg的溶液,作为对照溶液。杂质对照品溶液:取水杨酸、愈创木酚及水杨酸愈创木酚酯对照品各适量,精密称定,分别加流动相溶解并稀释制成每1ml中分别含水杨酸2μg、愈创木酚2μg及水杨酸愈创木酚酯4μg的溶液,作为杂质对照品溶液。操作方法:取对照溶液5μl注入液相色谱仪,记录色谱图,调节检测灵敏度,使主成分色谱峰的峰高约为满量程的20%-25%。记录色谱图,理论板数按呱西替柳峰计算应不低于2500。出峰顺序为水杨酸、愈创木酚及水杨酸愈创木酚酯,三者的分离度应符合要求。再分别量取供试品溶液、对照溶液及杂质对照品溶液各20μl,分别注入液相色谱仪,记录色谱图。检测结果:见表4表4有关物质检测结果总杂(%)单杂(%)得到的呱西替柳的晶型hplc图谱对比11.330.641a图3(结果分析见表5)对比30.990.519a图4(结果分析见表6)对比41.120.599a图5(结果分析见表7)对比50.760.406a图6(结果分析见表8)实施例10.190.079b图7(结果分析见表9)实施例20.170.078b图8(结果分析见表10)表5对比1有关物质检测结果分析保留时间(分钟)面积(微伏*秒)高度(微伏)usp理论塔板数%面积11.67933301088109060.02027.4441702184114260.010310.5441615552713033951680098.668415.0863004180183180.018515.8515754307143880.035620.3904516209130590.028721.196835573336162780.510824.8381049833954194510.641943.94211250241183280.069表6对比3有关物质检测结果分析保留时间(分钟)面积(微伏*秒)高度(微伏)usp理论塔板数%面积11.67258223668318940.03627.1373080363145330.019310.0671608987813648331691699.007414.9893865237114860.024519.828642742750164570.396623.510843163368200620.519表7对比4有关物质检测结果分析保留时间(分钟)面积(微伏*秒)高度(微伏)usp理论塔板数%面积17.0953660414143270.020210.0091796620515223451670598.880313.98117671570.010414.18639902430.022514.8766885385137830.038618.7464667216136620.026719.6111088944643161470.599823.325561212220188820.309940.08517569346115490.097表8对比5有关物质检测结果分析保留时间(分钟)面积(微伏*秒)高度(微伏)usp理论塔板数%面积11.7283971134892030.023210.0641712559514469251673199.243319.759701103146169700.406423.449564892370212950.327表9实施例1有关物质检测结果分析保留时间(分钟)面积(微伏*秒)高度(微伏)usp理论塔板数%面积12.496125934873890.00927.270170216367330.012310.3101438044211667501578499.814411.15311101290.008515.46411628629037220.008620.63511409475163500.079724.22910210417182410.071表10实施例2有关物质检测结果分析保留时间(分钟)面积(微伏*秒)高度(微伏)usp理论塔板数%面积12.492163740795050.01127.2912125240151030.014310.3301493609612128391583999.827420.67010512443160920.070524.29911655452184650.078实验例4:稳定性实验取各对比文件和各实施例得到的样品(编号为样品1到样品8,详见表1),模拟上市包装,置于40℃/rh75%恒温恒湿箱内贮存,分别于第1、2、3、6个月取样检测,结果见表11。表11加速试验考察结果由表4、表11结果可以看出:本发明实施例1、2得到的呱西替柳稳定性更好;实施例3-5的检测结果与实施例1、2一致。而本发明实施例6采用已有的精制方法得到的呱西替柳其稳定性与现有技术无差别。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1