一种吩噁嗪化合物或吩噻嗪化合物的合成方法与流程
本发明涉及氧(硫)氮类化合物合成技术领域,具体涉及一类吩噁嗪和吩噻嗪衍生物及其制备方法。
背景技术:
吩噁嗪和吩噻嗪类衍生物是一类非常重要的有机杂环化合物,因其衍生物具有抗癌、抗肿瘤、抗结核、抗菌、抗氧化等生物活性而广泛的应用于药物化学和生物化学的合成研究中[Prinz,H.;Chamasmani,B.;Vogel,K.;et al.J.Med.Chem.2011,54,4247.]。此外,吩噁嗪和吩噻嗪所特有的给电子体结构使其在染料敏化太阳能电池[Lee,W.;Yuk,S.B.;Choi,J.;et al.Dyes and Pigments 2014,102,13.]、荧光染料[Duan,C.;Li,J.;Han,C.;et al.Chem.Mater.2016,28,5667.]、和OLEDs[Okamoto,T.;Kozaki,M.;Doe,M.;et al.Chem.Mater.2005,17,5504.]等光电材料方面扮演着重要的角色。通常合成吩噁嗪和吩噻嗪类化合物的方法是通过三步法,首先两步法以钯催化铜催化或碱催化分子间和分子内关环合成未取代吩噁嗪或吩噻嗪类化合物,然后以该类化合为底物与卤代烃发生分子间C-N构建反应目标衍生物。该类合成方法大多是基于Buchwald-Hartwig胺化反应,需要将胺源引入底物,然后通过分子内N位单芳基化反应的方式两步合成目标吩噁嗪或吩噻嗪类衍生物(见反应1,2)。实验数据证明,该类反应对底物要求高因此选择范围极窄,且需要分步反应关环,反应条件较为苛刻(如较高的反应温度,添加大过量的碱),官能团容忍性较差,尤其是胺源可选范围窄。
反应1
反应2
技术实现要素:
本发明提供了一种条件温和、环境友好且能通过钯催化双卤化合物与伯胺分子间关环来合成吩噁嗪(吩噻嗪)类化合物的新方法。该方法避免了将胺源先引入底物的步骤,实现了一锅煮合成。
实现本发明的技术方案是:一种吩噁嗪化合物或吩噻嗪化合物的合成方法,具体步骤如下:在氮气氛围下,将双卤化合物、伯胺、钯化合物、膦配体、碱溶解在有机溶剂中,加热搅拌反应,之后经萃取、干燥、浓缩、纯化,制得吩噁嗪化合物或吩噻嗪化合物;
所述吩噁嗪化合物或吩噻嗪化合物的通式如下:
通式中X为O或S;R1表示氢原子、烷基、芳基、酰基、卤基、杂环基或酯基;R2表示烷基、芳基、杂环基或环烷基。
所述双卤化合物、伯胺、钯化合物、膦配体、碱的摩尔比为双卤化合物:伯胺:钯化合物:膦配体:碱=(1-2):(1-1.1):(0.05-0.10):(0.10-0.20):(3-6)。
所述的加热搅拌反应温度为100-120℃;反应时间为15-72h。
所述双卤化合物的通式如下:
通式中X表示O或S;R1表示氢原子、烷基、芳基、酰基、卤基、杂环基或酯基。
所述伯胺的通式为:R2-NH2,通式中R2表示烷基、芳基、杂环基。
所述的钯化合物为Pd(OAc)2、Pd2(dba)3、PdCl2、Pd(PPh3)2Cl2或PdCl2(dppf)中的任意一种。
所述的膦配体为如下通式所示化合物L1-L8中的任一种:
所述的碱为叔丁醇钠、叔丁醇钾、碳酸铯、碳酸钾、碳酸钠、三乙胺、叔丁胺、1,8-二氮杂双环(5.4.0)十一-7-烯中的任意一种。
所述的有机溶剂为甲苯、1,4-二氧六环、N,N-二甲基甲酰胺、四氢呋喃中的任意一种。
本发明提出一种合成吩噁嗪(吩噻嗪)类化合物的新思路,即通过钯催化双卤化合物与伯胺双胺化找到一种条件温和、环境友好且能通过仅一步反应高效快捷合成吩噁嗪(吩噻嗪)类化合物的新方法(见反应3)。该合成路径避免了将胺源事先引入底物,不仅能极大拓展胺源范围,也大范围拓展了底物官能团的容忍性,且反应条件更加温和,对于以往传统方法不能够合成吩噁嗪(吩噻嗪)类化合物也有较好的产率。该种一锅煮的新合成方法极大地节省了反应工作量,且反应过程中很少有副产物的生成,使反应的后处理更为便捷、高效。
反应3
本发明的有益效果是:本发明合成步骤简单、反应条件温和、操作简便易行、产物收率高达99%,而且所适用的底物范围广泛,为吩噁嗪(吩噻嗪)类化合物的合成提供了一种新思路,在有机合成方法学上具有重大的意义。
附图说明
图1是实施例1制备的目标产物A的1H NMR图。
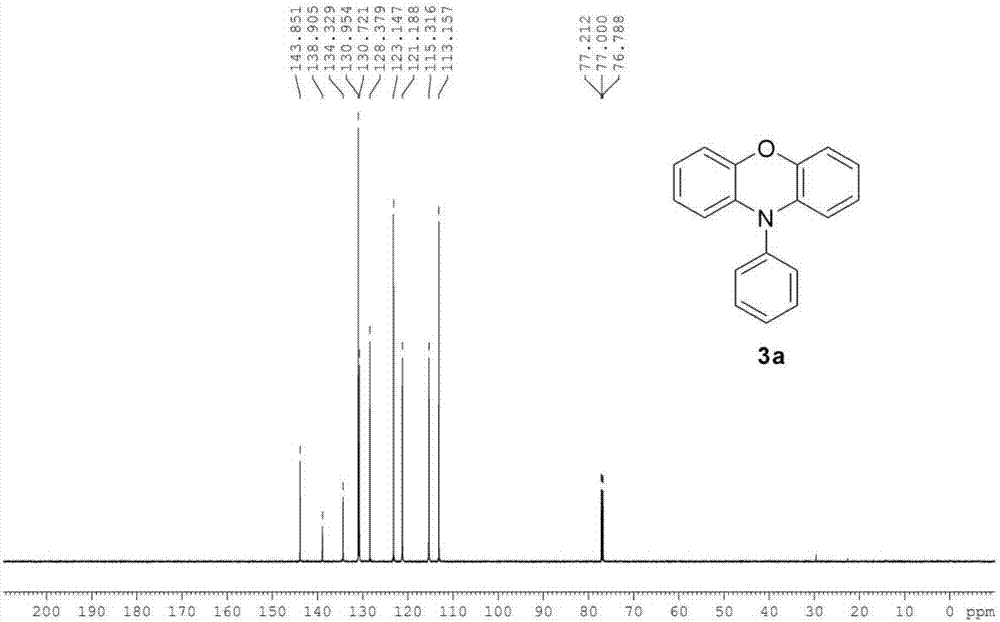
图2是实施例1制备的化合物A的13C NMR。
图3是实施例6制备的化合物F的1H NMR。
图4是实施例6制备的化合物F的13C NMR。
具体实施方式
下面结合实例来进一步描述本发明,通过下述实施例有助于进一步理解本发明,但并不限制本发明的范围。
实施例1
结构式如下的吩噁嗪类化合物A的制备方法:
向干燥的25mL Schlenk反应管中加入双卤化合物2,2’-二溴二苯醚(0.2mmol),然后依次加入苯胺(0.22mmol)、Pd(OAc)2(0.01mmol,5mol%)、DPEphos(0.02mmol,10mol%)、叔丁醇钠(0.6mmol),在氮气保护下,加入甲苯(2mL),120℃反应15h。反应结束后,经萃取,干燥,浓缩,柱色谱分离(纯石油醚)得到目标产物A。
1H NMR(600MHz,CDCl3,SiMe4):δ=7.53(t,J=7.74Hz,2H),7.41(t,J=7.38Hz,1H),7.29(d,J=7.86Hz,2H),6.66-6.65(m,2H),6.59(t,J=7.68Hz,2H),6.54-6.52(m,2H),5.87(d,J=7.98Hz,2H)。
13C NMR(150MHz,CDCl3,SiMe4):δ=143.85,138.90,134.33,130.95,130.72,128.38,123.15,121.19,115.32,113.16。
实施例2
结构式如下的吩噁嗪类化合物B的制备方法:
向干燥的25mL Schlenk反应管中加入双卤化合物2,2’-二溴二苯醚(1.2mmol),然后依次加入对苯二胺)(0.6mmol)、Pd(OAc)2(0.06mmol,10mol%)、DPEphos(0.12mmol,20mol%)、叔丁醇钠(3.6mmol),在氮气保护下,加入甲苯(2mL),120℃反应72h。反应结束后,经萃取,干燥,浓缩,柱色谱分离(石油醚:乙酸乙酯=20:1,体积比)得到目标产物B。
实施例3
结构式如下的吩噁嗪类化合物C的制备方法:
向干燥的25mL Schlenk反应管中加入双卤化合物2-溴-1-(2-溴苯氧基-4-氯)-苯(0.2mmol),然后依次加入苯胺(0.22mmol)、Pd(OAc)2(0.01mmol,5mol%)、DPEphos(0.02mmol,10mol%)、叔丁醇钠(0.6mmol),在氮气保护下,加入甲苯(2mL),120℃反应15h。反应结束后,经萃取,干燥,浓缩,柱色谱分离(纯石油醚)得到目标产物C。
实施例4
结构式如下的吩噻嗪类化合物D的制备方法:
向干燥的25mL Schlenk反应管中加入双卤化合物2,2'-二溴二苯硫醚(0.2mmol),然后依次加入苯胺(0.22mmol)、Pd2(dba)3(0.01mmol,5mol%)、BINAP(0.02mmol,10mol%)、叔丁醇钠(0.6mmol),在氮气保护下,加入甲苯(2mL),110℃反应20h。反应结束后,经萃取,干燥,浓缩,柱色谱分离(纯石油醚)得到目标产物D。
实施例5
结构式如下的吩噁嗪类化合物E的制备方法:
向干燥的25mL Schlenk反应管中加入双卤化合物2,2’-二溴二苯醚(0.2mmol),然后依次加入正辛胺(0.22mmol)、Pd(OAc)2(0.01mmol,5mol%)、BINAP(0.02mmol,10mol%)、叔丁醇钠(0.6mmol),在氮气保护下,加入甲苯(2mL),100℃反应24h。反应结束后,经萃取,干燥,浓缩,柱色谱分离(纯石油醚)得到目标产物E。
实施例6
结构式如下的吩噻嗪类化合物F的制备方法:
向干燥的25mL Schlenk反应管中加入双卤化合物2,2’-二溴二苯硫醚(1.2mmol),然后依次加入对苯二胺(0.6mmol)、Pd2(dba)3(0.06mmol,10mol%)、BINAP(0.12mmol,20mol%)、叔丁醇钠(3.6mmol),在氮气保护下,加入甲苯(2mL),110℃反应72h。反应结束后,经萃取,干燥,浓缩,柱色谱分离(石油醚:乙酸乙酯=20:1,体积比)得到目标产物F。
1H NMR(400MHz,CDCl3,SiMe4):δ=7.48(s,4H),7.11(d,J=7.28Hz,4H),7.01-6.88(m,8H),6.51(d,J=8.00Hz,4H)。
13C NMR(100MHz,CDCl3,SiMe4):δ=143.74,140.35,130.35,127.21,126.98,123.23,122.79,117.86。
不同条件下制备吩噁嗪或吩噻嗪化合物见表1。
表1不同条件下制备吩噁嗪或吩噻嗪化合物
注:a表示下列物质的摩尔比,即双卤化合物:伯胺:钯化合物:膦配体:碱
以上所述均为本发明的部分实施例,并非用来限制本发明。但凡依本发明内容所做的均等变化与修饰,都为本发明的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!