一种降血脂药物环丙贝特的合成方法与流程
本发明属于药物合成技术领域,具体涉及一种降血脂药物环丙贝特的合成方法。
背景技术:
贝特类药物又称苯氧芳酸类药物,是指包括吉非贝齐、氯贝特、非诺贝特、苯扎贝特和环丙贝特等在内的一类调脂药物。这类药物口服吸收快而完全,服药后1~2h内即可检测到血浆中药物浓度,与血浆蛋白结合率高。贝特类药物的半衰期从数小时至24h不等。环丙贝特有降血脂作用,通过改善胆固醇的分布,可减少ch和ldl在血管壁的沉积。
专利cn201310237441.9以对羟基苯乙烯为起始物料,经由bargllini反应一步构建羧基端,再经烯烃的卡宾插入反应构建三元环。缺点:原料对羟基苯乙烯不易经商业途径获得且性质不稳定,常作为中间体;另外两步反应都需引入过量强碱,反应条件不够温和。
专利cn201310590127.9报道了一种合成环丙贝特的路线。苯乙烯先在强碱条件下经由二氯卡宾插入反应构建三元环,再经friedel-crafts酰基化反应、baeyer-villiger氧化-醇解在对位引入羟基,最后再经烃化、水解得环丙贝特。缺点:虽反应原料简单,条件相对温和,但反应路线冗长,降低了收率,且原料苯乙烯性质不够稳定(易自聚),限制其应用。
技术实现要素:
本部分的目的在于概述本发明的实施例的一些方面以及简要介绍一些较佳实施例。在本部分以及本申请的说明书摘要和发明名称中可能会做些简化或省略以避免使本部分、说明书摘要和发明名称的目的模糊,而这种简化或省略不能用于限制本发明的范围。
鉴于上述和/或现有环丙贝特制备方法中存在的问题,提出了本发明。
因此,本发明目的是改变环丙贝特的合成路线,提供一种原子经济更高的降血脂药物环丙贝特的合成方法。
为解决上述技术问题,本发明提供了如下技术方案:一种降血脂药物环丙贝特的合成方法,包括,对羟基苯甲醛与丙二酸在混合溶剂中经碱催化生成对羟基苯乙烯;对羟基苯乙烯与丙酮、氯仿、碱经相转移催化剂催化生成中间体2-甲基-2-(4-乙烯基苯氧基)丙酸;2-甲基-2-(4-乙烯基苯氧基)丙酸与ticl4、mg、ccl4作用,生成环丙贝特。
作为本发明所述降血脂药物环丙贝特的合成方法的一种优选方案,其中:所述对羟基苯甲醛,其与丙二酸的摩尔比为1:1~4。
作为本发明所述降血脂药物环丙贝特的合成方法的一种优选方案,其中:所述混合溶剂包括dmf和/或甲苯。
作为本发明所述降血脂药物环丙贝特的合成方法的一种优选方案,其中:所述在混合溶剂中经碱催化生成对羟基苯乙烯,包括,碱催化反应先在甲苯中进行,反应1~2小时后冷却,将油状产物分出,再加入dmf继续升温反应。
作为本发明所述降血脂药物环丙贝特的合成方法的一种优选方案,其中:所述对羟基苯乙烯与丙酮、氯仿、碱,其中,碱包括koh、naoh、甲醇钠或乙醇钠中的一种或几种,其与对羟基苯乙烯摩尔比为4~6:1。
作为本发明所述降血脂药物环丙贝特的合成方法的一种优选方案,其中:所述对羟基苯乙烯与丙酮的摩尔比为1:1~5,所述对羟基苯乙烯与氯仿的摩尔比为1:1~5。
作为本发明所述降血脂药物环丙贝特的合成方法的一种优选方案,其中:所述相转移催化剂包括四正丁基溴化铵,其与对羟基苯乙烯的摩尔比为1:5~20。
作为本发明所述降血脂药物环丙贝特的合成方法的一种优选方案,其中:所述对羟基苯乙烯与丙酮、氯仿、碱经相转移催化剂催化生成中间体2-甲基-2-(4-乙烯基苯氧基)丙酸,其反应温度为10~60℃。
作为本发明所述降血脂药物环丙贝特的合成方法的一种优选方案,其中:所述2-甲基-2-(4-乙烯基苯氧基)丙酸与ticl4、mg、ccl4作用,其中,所述2-甲基-2-(4-乙烯基苯氧基)丙酸与ticl4摩尔比为1:0.1~1;所述2-甲基-2-(4-乙烯基苯氧基)丙酸与mg摩尔比为1:0.5~3;所述2-甲基-2-(4-乙烯基苯氧基)丙酸与ccl4摩尔比为1:0.5~3。
作为本发明所述降血脂药物环丙贝特的合成方法的一种优选方案,其中:所述2-甲基-2-(4-乙烯基苯氧基)丙酸与ticl4、mg、ccl4作用,其中,反应溶剂包括体积比为1:0.05~1的dcm/thf混合溶剂,反应温度为0℃~40℃,搅拌速度为100~200rpm。
本发明所具有的有益效果:
1、本发明反应路线短,原料廉价易得,反应条件温和,减少了能耗,降低了生产成本;后处理步骤简单,减少了三废的排放,绿色安全;相比于前人发表的路线,本路线的反应选择性与转化更高,三步反应的总转化率达81.2%,减少了原料的浪费,有较强的经济性。
2、产品杂质易分离,纯化后纯度达到98.6%。
附图说明
为了更清楚地说明本发明实施例的技术方案,下面将对实施例描述中所需要使用的附图作简单地介绍,显而易见地,下面描述中的附图仅仅是本发明的一些实施例,对于本领域普通技术人员来讲,在不付出创造性劳动性的前提下,还可以根据这些附图获得其它的附图。其中:
图1为本发明环丙贝特的合成路线;
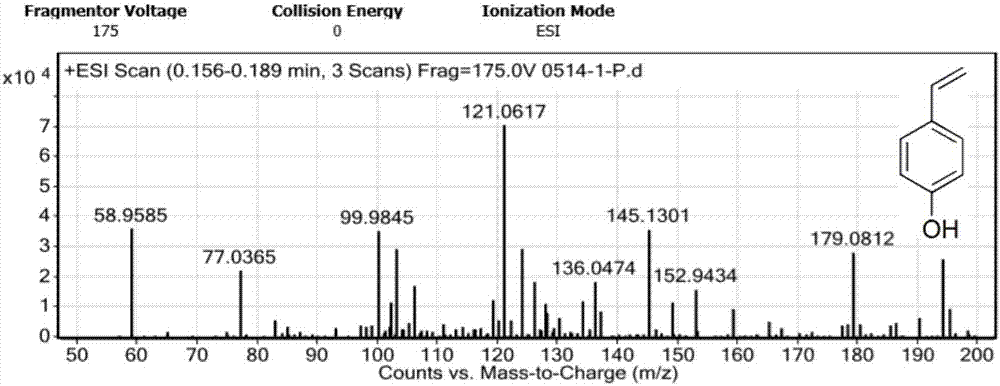
图2为中间产物对羟基苯乙烯(ii)的esi-ms谱图(正离子),图中m/z=121.06处为[m+h]+峰,结合图3共同验证了产物(ii)的正确;
图3为中间产物对羟基苯乙烯(ii)的1hnmr谱图(300mhz,chloroform-d)δ7.31~7.29(d,2h),6.80~6.77(d,2h),6.70~6.60(q,1h),5.62~5.56(d,1h),5.14~5.11(d,1h),4.71(s,1h).结合图2共同验证了产物(ii)的正确;
图4为中间产物2-甲基-2-(4-乙烯基苯氧基)丙酸(iii)的esi-ms谱图(正离子),图中m/z=229.1处为[m+na]+峰,结合图5共同验证了产物(iii)的正确;
图5为中间产物2-甲基-2-(4-乙烯基苯氧基)丙酸(iii)的1hnmr谱图(400mhz,chloroform-d)δ7.34~7.28(br,2h),6.91~6.87(br,2h),6.70~6.63(q,1h),5.66~5.62(d,1h),5.19~5.16(d,1h),1.63(s,6h).结合图4共同验证了产物(iii)的正确;
图6为产物环丙贝特(iv)的esi-ms谱图(负离子),图中m/z=287.0处为[m-h]-峰,结合图7共同验证了产物环丙贝特的正确;
图7为产物环丙贝特(iv)的1hnmr谱图(300mhz,chloroform-d)δ7.18~7.14(br,2h),6.93~6.90(br,2h),2.87~2.83(t,1h),1.98~1.94(t,1h),1.84~1.77(t,1h),1.61(s,6h).结合图6共同验证了产物环丙贝特的正确。
具体实施方式
为使本发明的上述目的、特征和优点能够更加明显易懂,下面结合具体实施例对本发明的具体实施方式做详细的说明。
在下面的描述中阐述了很多具体细节以便于充分理解本发明,但是本发明还可以采用其他不同于在此描述的其它方式来实施,本领域技术人员可以在不违背本发明内涵的情况下做类似推广,因此本发明不受下面公开的具体实施例的限制。
其次,此处所称的“一个实施例”或“实施例”是指可包含于本发明至少一个实现方式中的特定特征、结构或特性。在本说明书中不同地方出现的“在一个实施例中”并非均指同一个实施例,也不是单独的或选择性的与其他实施例互相排斥的实施例。
本发明提供了一种环丙贝特的合成方法,该路线以对羟基苯甲醛为原料,经knoevenagel-脱羧反应、bargllini反应及烯烃插入反应制得最终产物。在knoevenagel-脱羧连串反应中,采用混合溶剂替代传统的单一溶剂,有效地降低了反应时间,促进了中间产物的转化;在烯烃插入反应中,采用ticl4/mg/ccl4替代传统的卡宾加成体系来构建三元环,避免了强碱的使用,反应在较低温度下进行,条件温和,降低了能耗。具体步骤包括:
(1)对羟基苯甲醛(i),与丙二酸在混合溶剂中经加热及碱催化生成对羟基苯乙烯(ii);
(2)对羟基苯乙烯(ii)与丙酮、氯仿、碱经相转移催化剂催化生成中间体2-甲基-2-(4-乙烯基苯氧基)丙酸(iii);
(3)2-甲基-2-(4-乙烯基苯氧基)丙酸(iii)与ticl4、mg、ccl4作用,生成环丙贝特。
试剂名称缩写为:
thf:四氢呋喃
dmf:n,n-二甲基甲酰胺
dcm:二氯甲烷
dbu:1,8-二氮杂二环十一碳-7-烯
实施例1
(1)对羟基苯乙烯(ii)的合成
将50g对羟基苯甲醛(i)溶于250ml甲苯中,加入85g丙二酸与41.5ml二乙胺,150℃下加热回流,分水器分水。反应2hr后,将油状产物分出,再加入250mldmf,继续升温至150℃,反应2hr,反应液冷却,冰浴条件下用1mol/l稀盐酸调节ph至3~4,乙酸乙酯萃取,有机相经饱和nahco3溶液洗涤,饱和nacl溶液洗涤,无水na2so4干燥,减压蒸去溶剂,得粗产品对羟基苯乙烯(ii)油状物46.0g,收率94.6%。
(2)2-甲基-2-(4-乙烯基苯氧基)丙酸(iii)的合成
将10g对羟基苯乙烯(ii)溶于70ml丙酮中,加入9.9gnaoh、2.6g四正丁基溴化铵,40℃下加热使其稳定回流;将29.8g氯仿溶于约100ml丙酮中,逐滴缓慢加入到反应液中,滴加过程持续1hr。向反应液中加入100ml二氯甲烷和200ml水萃取,水相用1mol/l稀盐酸调节ph至1,乙酸乙酯萃取,饱和nacl溶液洗涤,无水na2so4干燥,减压蒸去溶剂,正己烷重结晶,得2-甲基-2-(4-乙烯基苯氧基)丙酸(iii)白色固体15.7g,收率91.6%。
(3)环丙贝特(iv)的合成
在0℃下将5g2-甲基-2-(4-乙烯基苯氧基)丙酸(iii)溶于40mldcm+4mlthf中,加入0.57g镁粉、3.67gccl4、0.45gticl4,0℃恒温以120r/min的速度搅拌反应2hr后,将反应液倒入100ml饱和na2so4溶液中,乙酸乙酯萃取,有机相经饱和nacl溶液洗涤,活性炭脱色,无水na2so4干燥,减压蒸去溶剂,正己烷重结晶,得环丙贝特(iv)白色固体6.65g,收率94.8%。
三步反应总收率为82.1%,粗产品可通过活性炭脱色、正己烷重结晶的方法进行纯化,所得环丙贝特白色晶体的hplc纯度达98.6%。
实施例2
(1)对羟基苯乙烯(ii)的合成
将50g对羟基苯甲醛(i)溶于500ml1:1(v/v)dmf/甲苯中,加入85g丙二酸与41.5ml二乙胺,150℃下加热回流,分水器分水。反应4hr后,反应液冷却,冰浴条件下用1mol/l稀盐酸调节ph至3~4,乙酸乙酯萃取,有机相经饱和nahco3溶液洗涤,饱和nacl溶液洗涤,无水na2so4干燥,减压蒸去溶剂,得粗产品对羟基苯乙烯(ii)油状物46.0g,收率93.5%。
(2)2-甲基-2-(4-乙烯基苯氧基)丙酸(iii)的合成
将10g对羟基苯乙烯(ii)溶于70ml丙酮中,加入9.9gnaoh、2.6g四正丁基溴化铵,40℃下加热使其稳定回流;将29.8g氯仿溶于约100ml丙酮中,逐滴缓慢加入到反应液中,滴加过程持续1hr。向反应液中加入100ml二氯甲烷和200ml水萃取,水相用1mol/l稀盐酸调节ph至1,乙酸乙酯萃取,饱和nacl溶液洗涤,无水na2so4干燥,减压蒸去溶剂,正己烷重结晶,得2-甲基-2-(4-乙烯基苯氧基)丙酸(iii)白色固体15.7g,收率91.6%。
(3)环丙贝特(iv)的合成
在0℃下将5g2-甲基-2-(4-乙烯基苯氧基)丙酸(iii)溶于40mldcm+4mlthf中,加入0.57g镁粉、3.67gccl4、0.45gticl4,0℃恒温以120r/min的速度搅拌反应2hr后,将反应液倒入100ml饱和na2so4溶液中,乙酸乙酯萃取,有机相经饱和nacl溶液洗涤,活性炭脱色,无水na2so4干燥,减压蒸去溶剂,正己烷重结晶,得环丙贝特(iv)白色固体6.65g,收率94.8%。
三步反应总收率为81.2%,粗产品可通过活性炭脱色、正己烷重结晶的方法进行纯化,所得环丙贝特白色晶体的hplc纯度达98.6%。
实施例3
(1)对羟基苯乙烯(ii)的合成
将50g对羟基苯甲醛(i)溶于500ml1:1(v/v)dmf/甲苯中,加入85g丙二酸与41.5ml二乙胺,150℃下加热回流,分水器分水。反应4hr后,反应液冷却,冰浴条件下用1mol/l稀盐酸调节ph至3~4,乙酸乙酯萃取,有机相经饱和nahco3溶液洗涤,饱和nacl溶液洗涤,无水na2so4干燥,减压蒸去溶剂,得粗产品对羟基苯乙烯(ii)油状物46.0g,收率93.5%。
(2)2-甲基-2-(4-乙烯基苯氧基)丙酸(iii)的合成
将10g对羟基苯乙烯(ii)溶于70ml丙酮中,加入9.9gnaoh、2.6g四正丁基溴化铵,20℃下加热使其稳定回流;将29.8g氯仿溶于约100ml丙酮中,逐滴缓慢加入到反应液中,滴加过程持续1hr。向反应液中加入100ml二氯甲烷和200ml水萃取,水相用1mol/l稀盐酸调节ph至1,乙酸乙酯萃取,饱和nacl溶液洗涤,无水na2so4干燥,减压蒸去溶剂,正己烷重结晶,得2-甲基-2-(4-乙烯基苯氧基)丙酸(iii)白色固体15.5g,收率91.0%。
(3)环丙贝特(iv)的合成
在0℃下将5g2-甲基-2-(4-乙烯基苯氧基)丙酸(iii)溶于40mldcm+4mlthf中,加入0.57g镁粉、3.67gccl4、0.45gticl4,0℃恒温以120r/min的速度搅拌反应2hr后,将反应液倒入100ml饱和na2so4溶液中,乙酸乙酯萃取,有机相经饱和nacl溶液洗涤,活性炭脱色,无水na2so4干燥,减压蒸去溶剂,正己烷重结晶,得环丙贝特(iv)白色固体6.65g,收率94.8%。
三步反应总收率为80.7%,粗产品可通过活性炭脱色、正己烷重结晶的方法进行纯化,所得环丙贝特白色晶体的hplc纯度达98.6%。
实施例4
(1)对羟基苯乙烯(ii)的合成
将50g对羟基苯甲醛(i)溶于500ml1:1(v/v)dmf/甲苯中,加入85g丙二酸与41.5ml二乙胺,150℃下加热回流,分水器分水。反应4hr后,反应液冷却,冰浴条件下用1mol/l稀盐酸调节ph至3~4,乙酸乙酯萃取,有机相经饱和nahco3溶液洗涤,饱和nacl溶液洗涤,无水na2so4干燥,减压蒸去溶剂,得粗产品对羟基苯乙烯(ii)油状物46.0g,收率93.5%。
(2)2-甲基-2-(4-乙烯基苯氧基)丙酸(iii)的合成
将10g对羟基苯乙烯(ii)溶于70ml丙酮中,加入9.9gnaoh、2.6g四正丁基溴化铵,40℃下加热使其稳定回流;将29.8g氯仿溶于约100ml丙酮中,逐滴缓慢加入到反应液中,滴加过程持续1hr。向反应液中加入100ml二氯甲烷和200ml水萃取,水相用1mol/l稀盐酸调节ph至1,乙酸乙酯萃取,饱和nacl溶液洗涤,无水na2so4干燥,减压蒸去溶剂,正己烷重结晶,得2-甲基-2-(4-乙烯基苯氧基)丙酸(iii)白色固体15.7g,收率91.6%。
(3)环丙贝特(iv)的合成
在40℃下将5g2-甲基-2-(4-乙烯基苯氧基)丙酸(iii)溶于40mldcm+4mlthf中,加入0.57g镁粉、3.67gccl4、0.45gticl4,40℃恒温以200r/min的速度搅拌反应2hr后,将反应液倒入100ml饱和na2so4溶液中,乙酸乙酯萃取,有机相经饱和nacl溶液洗涤,活性炭脱色,无水na2so4干燥,减压蒸去溶剂,正己烷重结晶,得环丙贝特(iv)白色固体6.54g,收率93.2%。
三步反应总收率为81.2%,粗产品可通过活性炭脱色、正己烷重结晶的方法进行纯化,所得环丙贝特白色晶体的hplc纯度达98.6%。
实施例5
(1)对羟基苯乙烯(ii)的合成
将50g对羟基苯甲醛(i)溶于500ml1:1(v/v)dmf/甲苯中,加入85g丙二酸与62.8mldbu,150℃下加热回流,分水器分水。反应4hr后,反应液冷却,冰浴条件下用1mol/l稀盐酸调节ph至3~4,乙酸乙酯萃取,有机相经饱和nahco3溶液洗涤,饱和nacl溶液洗涤,无水na2so4干燥,减压蒸去溶剂,得粗产品对羟基苯乙烯(ii)油状物45.3g,收率92.1%。
(2)2-甲基-2-(4-乙烯基苯氧基)丙酸(iii)的合成
将10g对羟基苯乙烯(ii)溶于70ml丙酮中,加入11.6gkoh、2.6g四正丁基溴化铵,40℃下加热使其稳定回流;将19.9g氯仿溶于约100ml丙酮中,逐滴缓慢加入到反应液中,滴加过程持续1hr。向反应液中加入100ml二氯甲烷和200ml水萃取,水相用1mol/l稀盐酸调节ph至1,乙酸乙酯萃取,饱和nacl溶液洗涤,无水na2so4干燥,减压蒸去溶剂,正己烷重结晶,得2-甲基-2-(4-乙烯基苯氧基)丙酸(iii)白色固体14.8g,收率86.3%。
(3)环丙贝特(iv)的合成
在0℃下将5g2-甲基-2-(4-乙烯基苯氧基)丙酸(iii)溶于40mldcm+4mlthf中,加入0.29g镁粉、4.40gccl4、0.23gticl4,0℃恒温以120r/min的速度搅拌反应2hr后,将反应液倒入100ml饱和na2so4溶液中,乙酸乙酯萃取,有机相经饱和nacl溶液洗涤,活性炭脱色,无水na2so4干燥,减压蒸去溶剂,正己烷重结晶,得环丙贝特(iv)白色固体5.86g,收率83.5%。
三步反应总收率为66.4%。
由此可见,本发明反应路线短,原料廉价易得,反应条件温和,减少了能耗,降低了生产成本;后处理步骤简单,减少了三废的排放,绿色安全;相比于前人发表的路线,本路线的反应选择性与转化更高,三步反应的总转化率达81.2%,减少了原料的浪费,有较强的经济性;产品杂质易分离,纯化后纯度达到98.6%。
应说明的是,以上实施例仅用以说明本发明的技术方案而非限制,尽管参照较佳实施例对本发明进行了详细说明,本领域的普通技术人员应当理解,可以对本发明的技术方案进行修改或者等同替换,而不脱离本发明技术方案的精神和范围,其均应涵盖在本发明的权利要求范围当中。
- 还没有人留言评论。精彩留言会获得点赞!