咔唑和膦氧双修饰均配位苯基苯并咪唑铱配合物、制备方法及其应用与流程
本发明属于有机电子功能材料技术领域,具体涉及一类咔唑和膦氧双修饰均配位苯基苯并咪唑铱配合物、制备方法及其在有机电致磷光器件中的应用。
背景技术:
有机电致发光二极管(oleds)具有超薄、超轻、响应快、发光效率高以及可挠曲等优点,在新一代的平板显示技术和照明领域引起广泛重视和研究。1997年
然而,电致磷光材料相对较长的激发态寿命容易导致浓度猝灭和三重态湮灭,从而极大地降低器件的发光效率和亮度。同时,小分子电致磷光配合物的溶解性较差,难以溶液加工成膜,在大尺寸和可图形化上处于明显劣势。针对以上问题,通常采用的方法是将磷光发光体(客体材料)掺杂或共混到主体基质中。然而,主客体之间的能量传递效率和相分离以及客体分子自聚集等都会导致器件效率和寿命的降低。因此,主客体一体化的概念被提出来,即将具有主体材料特点的功能基团通过共价键连接到重金属配合物上,进而避免了主客体相分离的发生。此类材料典型代表是树枝状电致磷光材料和电致磷光聚合物,它们不仅能够有效地分散发光中心,实现良好的能量传递,而且具有溶液加工特性,能够通过旋涂、喷墨打印和丝网印刷等溶液加工技术形成高质量的薄膜。但是它们的合成难度较大,材料质量不稳定。
技术实现要素:
本发明是要解决现有电致磷光材料相对较长的激发态寿命容易导致浓度猝灭和三重态湮灭的技术问题,提供了一种咔唑和膦氧双修饰均配位苯基苯并咪唑铱配合物、制备方法及其应用。
咔唑和膦氧双修饰均配位苯基苯并咪唑铱配合物中心离子为三价铱离子,由苯基苯并咪唑类碳氮配体经配合反应制得,其结构式如下:
(1)当x为三苯基膦氧,y为3,6-二叔丁基咔唑,在苯环的2,3位上取代时,化合物为ir(23dcznpopbi)3,其结构式为:
(2)当x为三苯基膦氧,y为3,6-二叔丁基咔唑,在苯环的2,4位上取代时,化合物为ir(24dcznpopbi)3,其结构式为:
(3)当x为三苯基膦氧,y为3,6-二叔丁基咔唑,在苯环的3,4位上取代时,化合物为ir(34dcznpopbi)3,其结构式为:
咔唑和膦氧双修饰均配位苯基苯并咪唑铱配合物的制备方法如下:
一、将1mmol的(4-羟基苯基)二苯基膦氧、0.1~0.5mmol的tbab、2~4mmol的koh、20~50ml的1,3-二氯丙烷在90℃条件下搅拌反应5~24小时,然后用水和ch2cl2萃取,合并有机相,用无水硫酸钠干燥,以石油醚和乙酸乙酯的混合溶剂为淋洗剂柱层析纯化,得到(4-3-氯丙氧基)苯基二苯基膦氧;
二、将1mmol的二羟基苯基苯并咪唑、0.1~0.5mmol的tbab、2~4mmol的koh、2~5mmol的9-(3-氯丙基)叔丁基咔唑溶解于10~50ml四氢呋喃中,于70℃条件下搅拌反应12~24小时,然后用水和ch2cl2萃取,合并有机相,用无水硫酸钠干燥,以石油醚和乙酸乙酯的混合溶剂为淋洗剂柱层析纯化,得到2,3/2,4/3,4-二叔丁基咔唑亚丙氧基苯基苯并咪唑;
三、将(4-3-氯丙氧基)苯基二苯基膦氧与2,3/2,4/3,4-二叔丁基咔唑亚丙氧基苯基苯并咪唑溶解于四氢呋喃中,加入0.1~0.5当量的tbab,1~2当量的nah,在70℃条件下搅拌反应12~24小时,然后用水和ch2cl2萃取,合并有机相,用无水硫酸钠干燥,以石油醚和乙酸乙酯的混合溶剂为淋洗剂柱层析纯化,得到2,3/2,4/3,4-二叔丁基咔唑亚丙氧基苯基-三苯基膦氧亚丙基苯并咪唑;
四、将2,3/2,4/3,4-二叔丁基咔唑亚丙氧基苯基-三苯基膦氧亚丙基苯并咪唑、0.5当量的三氯化铱、10~15ml的乙二醇独乙醚、10~15ml的水以及5~10ml的四氢呋喃加热至110℃搅拌反应12~24小时,然后冷却至室温,抽滤得到氯桥联中间体,将氯桥联中间体与1~2当量的2,3/2,4/3,4-二叔丁基咔唑亚丙氧基苯基-三苯基膦氧亚丙基苯并咪唑、2~5当量的k2co3以及10ml甘油加热至240℃搅拌反应12~48小时,反应结束后用水和ch2cl2萃取,合并有机相,用无水硫酸钠干燥,以石油醚和乙酸乙酯的混合溶剂为淋洗剂柱层析纯化,得到咔唑和膦氧双修饰均配位苯基苯并咪唑铱配合物。
所述咔唑和膦氧双修饰均配位苯基苯并咪唑铱配合物作为发光层材料用于有机电致磷光器件。
所述应用方法如下:
先制作导电层,然后在导电层上旋涂或喷墨打印空穴注入层材料,在空穴注入层上旋涂咔唑和膦氧双修饰均配位苯基苯并咪唑铱配合物或与咔唑和膦氧双修饰均配位苯基苯并咪唑铱配合物掺杂的掺杂体发光层,在发光层上蒸镀电子传输空穴阻挡材料,最后蒸镀第二层导电层。
所述掺杂体为tpxzpo与咔唑和膦氧双修饰均配位苯基苯并咪唑铱配合物掺杂。
本发明使用苯基苯并咪唑作为碳氮配体与铱离子形成重金属配合物核心。苯基苯并咪唑铱配合物是一类非常好的蓝绿光磷光材料,是绿光铱配合物中少数几个光致发光效率超过50%的。其次,本发明使用具有空穴传输能力的咔唑基团以及具有电子传输能力的膦氧基团作为外围修饰基团,一方面增强并平衡铱配合物的载流子传输,降低驱动电压;另一方面实现对发光中心的包封和隔离,有效降低猝灭效应,提高电致发光效率。最后,本发明使用饱和烷基将上述两部分连接起来。柔性的烷基能够提高配合物的溶解性;另外,外围咔唑基团上的叔丁基也可显著提高铱配合物的溶解性,更利于实现溶液加工特性。
相对于同类的苯基苯并咪唑铱配合物衍生物,本发明中使用的咔唑和膦氧双修饰均配位苯基苯并咪唑配体从根本上解决了三个影响配合物电致发光特性和加工性能的主要问题:
1.有效抑制浓度猝灭和三重态湮灭效应。咔唑和膦氧作为外围基团能够有效地包封和隔离苯基苯并咪唑铱配合物发光核,均匀地分散发光中心,降低发光核浓度和减少发光中心之间的相互作用;
2.提高并平衡铱配合物的载流子注入和传输能力,提高电致发光性能。外围的咔唑和膦氧基团分别具有良好的空穴和电子传输能力,可以增强配合物的载流子传输能力,增加发光层载流子复合概率,提高电致发光效率;
3.提高材料的溶液加工性能。本发明涉及的铱配合物含有大量的柔性基团,能够提供足够的溶解性,保证能够通过溶液加工方法制备高质量的薄膜。
咔唑膦氧双修饰均配位苯基苯并咪唑类配体不仅能够提高铱配合物的溶解性,在有效地降低猝灭效应的同时,提供良好的载流子注入和传输能力,从而提高铱配合物电致磷光器件的综合性能。在用于电致发光器件时,制成的器件在亮度和效率方面获得了令人满意的结果,其综合性能达到甚至优于目前已知的部分电致磷光铱聚合物和树枝状化合物,从而获得了一类具有广泛应用前景的光电功能铱配合物。
本发明提供的咔唑和膦氧双修饰均配位苯基苯并咪唑铱配合物以苯基苯并咪唑为碳氮配体,在其苯基和苯并咪唑的n位上以柔性链连接的方式引入具有载流子传输特性的咔唑和膦氧基团,从而实现九维混合修饰的均配位铱配合物。通过增加修饰位点和调控外围基团的空间分布,实现对发光中心的更为有效的包封和隔离,抑制发光中心的猝灭效应,从而提高电致发光效率和效率稳定性,简化器件结构。另外,咔唑上叔丁基的引入可以显著增强配合物的溶解性,更有利于通过溶液加工的方法制备电致发光器件。
附图说明
图1是实验一合成的配合物ir(23dcznpopbi)3的紫外荧光光谱谱图,其中■表示二氯甲烷溶剂中的紫外光谱图,●表示薄膜的紫外光谱图,□表示二氯甲烷溶剂中的荧光光谱图,○表示薄膜的荧光光谱图,△表示77k条件下的荧光光谱图;
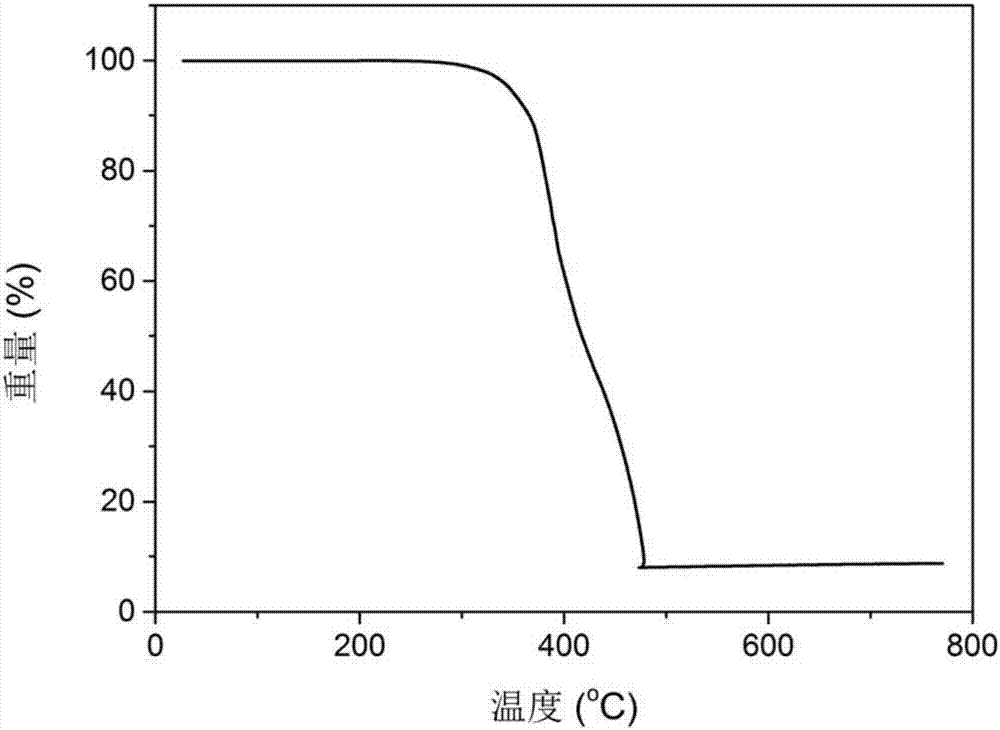
图2是实验一合成的配合物ir(23dcznpopbi)3的热重分析谱图;
图3是实验二合成的配合物ir(24dcznpopbi)3的紫外荧光光谱谱图,其中■表示二氯甲烷溶剂中的紫外光谱图,●表示薄膜的紫外光谱图,□表示二氯甲烷溶剂中的荧光光谱图,○表示薄膜的荧光光谱图,△表示77k条件下的荧光光谱图;
图4是实验二合成的配合物ir(24dcznpopbi)3的热重分析谱图;
图5是实验三合成的配合物ir(34dcznpopbi)3的紫外荧光光谱谱图,其中■表示二氯甲烷溶剂中的紫外光谱图,●表示薄膜的紫外光谱图,□表示二氯甲烷溶剂中的荧光光谱图,○表示薄膜的荧光光谱图,△表示77k条件下的荧光光谱图;
图6是实验三合成的配合物ir(34dcznpopbi)3的热重分析谱图;
图7是实验三以配合物ir(34dcznpopbi)3制备的掺杂型电致绿光磷光器件的电压-电流密度关系曲线;
图8是实验三以配合物ir(34dcznpopbi)3制备的掺杂型电致绿光磷光器件的电压-亮度关系曲线;
图9是实验三以配合物ir(34dcznpopbi)3制备的掺杂型电致绿光磷光器件的电流密度-电流效率关系曲线;
图10是实验三以配合物ir(34dcznpopbi)3制备的掺杂型电致绿光磷光器件的电流密度-功率效率关系曲线;
图11是实验三以配合物ir(34dcznpopbi)3制备的掺杂型电致绿光磷光器件的电流密度-外量子效率关系曲线;
图12是实验三以配合物ir(34dcznpopbi)3制备的掺杂型电致绿光磷光器件的电致发光光谱图;
图13是实验三以配合物ir(34dcznpopbi)3制备的非掺杂型电致绿光磷光器件的电压-电流密度关系曲线;
图14是实验三以配合物ir(34dcznpopbi)3制备的非掺杂型电致绿光磷光器件的电压-亮度关系曲线;
图15是实验三以配合物ir(34dcznpopbi)3制备的非掺杂型电致绿光磷光器件的电流密度-电流效率关系曲线;
图16是实验三以配合物ir(34dcznpopbi)3制备的非掺杂型电致绿光磷光器件的电流密度-功率效率关系曲线;
图17是实验三以配合物ir(34dcznpopbi)3制备的非掺杂型电致绿光磷光器件的电流密度-外量子效率关系曲线;
图18是实验三以配合物ir(34dcznpopbi)3制备的非掺杂型电致绿光磷光器件的电致发光光谱图。
具体实施方式
本发明技术方案不局限于以下所列举具体实施方式,还包括各具体实施方式间的任意组合。
具体实施方式一:本实施方式所述咔唑和膦氧双修饰均配位苯基苯并咪唑铱配合物中心离子为三价铱离子,由苯基苯并咪唑类碳氮配体经配合反应制得,其结构式如下:
(1)当x为三苯基膦氧,y为3,6-二叔丁基咔唑,在苯环的2,3位上取代时,化合物为ir(23dcznpopbi)3,其结构式为:
(2)当x为三苯基膦氧,y为3,6-二叔丁基咔唑,在苯环的2,4位上取代时,化合物为ir(24dcznpopbi)3,其结构式为:
(3)当x为三苯基膦氧,y为3,6-二叔丁基咔唑,在苯环的3,4位上取代时,化合物为ir(34dcznpopbi)3,其结构式为:
具体实施方式二:具体实施方式一所述咔唑和膦氧双修饰均配位苯基苯并咪唑铱配合物的制备方法,其特征在于该制备方法如下:
一、将1mmol的(4-羟基苯基)二苯基膦氧、0.1~0.5mmol的tbab、2~4mmol的koh、20~50ml的1,3-二氯丙烷在90℃条件下搅拌反应5~24小时,然后用水和ch2cl2萃取,合并有机相,用无水硫酸钠干燥,以石油醚和乙酸乙酯的混合溶剂为淋洗剂柱层析纯化,得到(4-3-氯丙氧基)苯基二苯基膦氧;
二、将1mmol的二羟基苯基苯并咪唑、0.1~0.5mmol的tbab、2~4mmol的koh、2~5mmol的9-(3-氯丙基)叔丁基咔唑溶解于10~50ml四氢呋喃中,于70℃条件下搅拌反应12~24小时,然后用水和ch2cl2萃取,合并有机相,用无水硫酸钠干燥,以石油醚和乙酸乙酯的混合溶剂为淋洗剂柱层析纯化,得到2,3/2,4/3,4-二叔丁基咔唑亚丙氧基苯基苯并咪唑;
三、将(4-3-氯丙氧基)苯基二苯基膦氧与2,3/2,4/3,4-二叔丁基咔唑亚丙氧基苯基苯并咪唑溶解于四氢呋喃中,加入0.1~0.5当量的tbab,1~2当量的nah,在70℃条件下搅拌反应12~24小时,然后用水和ch2cl2萃取,合并有机相,用无水硫酸钠干燥,以石油醚和乙酸乙酯的混合溶剂为淋洗剂柱层析纯化,得到2,3/2,4/3,4-二叔丁基咔唑亚丙氧基苯基-三苯基膦氧亚丙基苯并咪唑;
四、将2,3/2,4/3,4-二叔丁基咔唑亚丙氧基苯基-三苯基膦氧亚丙基苯并咪唑、0.5当量的三氯化铱、10~15ml的乙二醇独乙醚、10~15ml的水以及5~10ml的四氢呋喃加热至110℃搅拌反应12~24小时,然后冷却至室温,抽滤得到氯桥联中间体,将氯桥联中间体与1~2当量的2,3/2,4/3,4-二叔丁基咔唑亚丙氧基苯基-三苯基膦氧亚丙基苯并咪唑、2~5当量的k2co3以及10ml甘油加热至240℃搅拌反应12~48小时,反应结束后用水和ch2cl2萃取,合并有机相,用无水硫酸钠干燥,以石油醚和乙酸乙酯的混合溶剂为淋洗剂柱层析纯化,得到咔唑和膦氧双修饰均配位苯基苯并咪唑铱配合物。
具体实施方式三:本实施方式与具体实施方式二不同的是步骤一中将1mmol的(4-羟基苯基)二苯基膦氧、0.2~0.4mmol的tbab、3mmol的koh、30~40ml的1,3-二氯丙烷在90℃条件下搅拌反应10~20小时。其他与具体实施方式二相同。
具体实施方式四:本实施方式与具体实施方式二或三不同的是步骤二中将1mmol的二羟基苯基苯并咪唑、0.2~0.4mmol的tbab、3mmol的koh、4mmol的9-(3-氯丙基)叔丁基咔唑溶解于20~40ml四氢呋喃中,于70℃条件下搅拌反应18~20小时。其他与具体实施方式二或三相同。
具体实施方式五:本实施方式与具体实施方式二至四之一不同的是步骤三中加入0.4当量的tbab,1.5当量的nah,在70℃条件下搅拌反应20小时。其他与具体实施方式二至四之一相同。
具体实施方式六:本实施方式与具体实施方式二至五之一不同的是步骤四中将2,3/2,4/3,4-二叔丁基咔唑亚丙氧基苯基-三苯基膦氧亚丙基苯并咪唑、0.5当量的三氯化铱、12~13ml的乙二醇独乙醚、11~14ml的水以及6~8ml的四氢呋喃加热至110℃搅拌反应15~20小时。其他与具体实施方式二至三之一相同。
具体实施方式七:本实施方式与具体实施方式二至三之一不同的是步骤四中将2,3/2,4/3,4-二叔丁基咔唑亚丙氧基苯基-三苯基膦氧亚丙基苯并咪唑、0.5当量的三氯化铱、11ml的乙二醇独乙醚、13ml的水以及7ml的四氢呋喃加热至110℃搅拌反应18小时。其他与具体实施方式二至三之一相同。
具体实施方式八:具体实施方式一所述咔唑和膦氧双修饰均配位苯基苯并咪唑铱配合物作为发光层材料用于有机电致磷光器件。
具体实施方式九:本实施方式与具体实施方式八不同的是所述应用方法如下:
先制作导电层,然后在导电层上旋涂或喷墨打印空穴注入层材料,在空穴注入层上旋涂咔唑和膦氧双修饰均配位苯基苯并咪唑铱配合物或与咔唑和膦氧双修饰均配位苯基苯并咪唑铱配合物掺杂的掺杂体发光层,在发光层上蒸镀电子传输空穴阻挡材料,最后蒸镀第二层导电层。
第一层导电层制作在衬底上,衬底为玻璃或塑料。
第一层导电层为阳极,第二层导电层为阴极。阴极最好由能产生反射的金属或者半透明导体构成,一般为钙、镁、铝、银及其合金,使用铝时在铝表面镀上一层lif。阳极由氧化铟锡(ito)或透明导电聚合物(如聚苯胺,pani)构成。
在第一层导电层和发光层之间有一层空穴注入层,电子传输/空穴阻挡层处于第二层导电层与发光层之间;发光层介于空穴注入层与电子传输/空穴阻挡层之间。
本发明中的器件阳极由氧化铟锡(ito)或聚苯胺(pani)通过蒸镀到玻璃或塑料透明基质上制成。
阴极由氟化锂/铝(lif/al)、ba/al或镁银合金构成(厚度为10nm/1000nm)。
为了提高阳极的空穴注入和传输、以及阴极的电子注入和传输,并使电荷能在发光层有效地复合,可以采用空穴注入层和电子传输/空穴阻挡层。
空穴注入层薄膜的厚度为20~60nm,可以通过旋涂或喷墨打印在阳极上成膜。
发光层为纯净的铱配合物或者铱配合物与tpxzpo形成的掺杂体或共混体,薄膜厚度为10-100nm,可以通过溶液加工技术沉积到空穴注入层上。
电子传输/空穴阻挡层薄膜的厚度为10~80nm,通过热蒸发技术沉积到发光层上。
具体实施方式十:本实施方式与具体实施方式八或九不同的是所述掺杂体为tpxzpo与咔唑和膦氧双修饰均配位苯基苯并咪唑铱配合物掺杂。其他与具体实施方式八或九相同。
采用下述实验验证本发明效果:
实验一:本实验咔唑和膦氧双修饰均配位苯基苯并咪唑铱配合物ir(23dcznpopbi)3的合成方法按下列步骤实现:
一、将1mmol的2,3-二羟基苯基苯并咪唑、0.4mmol的tbab、3mmol的koh、4mmol的9-(3-氯丙基)叔丁基咔唑溶解于20ml四氢呋喃中70℃条件下搅拌反应15小时,然后用水和ch2cl2萃取,合并有机相,用无水硫酸钠干燥,以石油醚和乙酸乙酯的混合溶剂为淋洗剂柱层析纯化得到2,3-二叔丁基咔唑亚丙氧基苯基苯并咪唑;
二、将步骤一合成的2,3-二叔丁基咔唑亚丙氧基苯基苯并咪唑溶解于四氢呋喃中,加入1.5mmol的(4-3-氯丙氧基)苯基二苯基膦氧、0.4mmol的tbab,1.5mmol的nah,70℃条件下搅拌反应18小时,然后用水和ch2cl2萃取,合并有机相,用无水硫酸钠干燥,以石油醚和乙酸乙酯的混合溶剂为淋洗剂柱层析纯化得到2,3-二叔丁基咔唑亚丙氧基苯基-三苯基膦氧亚丙基苯并咪唑;
三、将步骤二合成的配体2,3-二叔丁基咔唑亚丙氧基苯基-三苯基膦氧亚丙基苯并咪唑、0.8mmol的三氯化铱、12ml的乙二醇独乙醚、12ml的水以及8ml的四氢呋喃加热至110℃搅拌反应20小时。然后冷却至室温,抽滤得到氯桥联中间体。将此中间体与1当量的2,3-二叔丁基咔唑亚丙氧基苯基-三苯基膦氧亚丙基苯并咪唑、3当量的k2co3以及10ml甘油加热至240℃搅拌反应24小时。反应结束后用水和ch2cl2萃取,合并有机相,用无水硫酸钠干燥,以石油醚和乙酸乙酯的混合溶剂为淋洗剂柱层析纯化得到铱配合物ir(23dcznpopbi)3。
本实验步骤一制备的2,3-二叔丁基咔唑亚丙氧基苯基苯并咪唑,其结构式为
按照实验一中步骤一制备的2,3-二叔丁基咔唑亚丙氧基苯基苯并咪唑,其飞行时间质谱的数据为:m/z(%):864(100)[m+];元素分析的数据为:分子式c59h68n4o2,理论值:c81.90,h7.92,n6.48,实测值:c81.95,h7.93,n6.48。
步骤二制备的2,3-二叔丁基咔唑亚丙氧基苯基-三苯基膦氧亚丙基苯并咪唑,其飞行时间质谱的数据为:m/z(%):1198(100)[m+];元素分析的数据为:分子式c80h87n4o4p,理论值:c80.10,h7.31,n4.67;实测值:c80.15,h7.30,n4.69。
步骤三制备的铱配合物ir(23dcznpopbi)3,其飞行时间质谱的数据为:m/z(%):3790(100)[m+];元素分析的数据为:分子式c240h261irn12o12p3,理论值:c76.04,h6.94,n4.43;实测值:c76.01,h6.97,n4.45。
本实验得到咔唑和膦氧双修饰均配位苯基苯并咪唑铱配合物ir(23dcznpopbi)3的紫外荧光光谱谱图如图1所示。
本实验得到咔唑和膦氧双修饰均配位苯基苯并咪唑铱配合物ir(23dcznpopbi)3的热重分析谱图如图2所示,由图可知铱配合物ir(23dcznpopbi)3的裂解温度达344℃。
实验二:本实验咔唑和膦氧双修饰均配位苯基苯并咪唑铱配合物ir(24dcznpopbi)3的合成方法按下列步骤实现:
一、将1mmol的2,4-二羟基苯基苯并咪唑、0.2mmol的tbab、3mmol的koh、3mmol的9-(3-氯丙基)叔丁基咔唑溶解于30ml四氢呋喃中70℃条件下搅拌反应20小时,然后用水和ch2cl2萃取,合并有机相,用无水硫酸钠干燥,以石油醚和乙酸乙酯的混合溶剂为淋洗剂柱层析纯化得到2,4-二叔丁基咔唑亚丙氧基苯基苯并咪唑;
二、将步骤一合成的2,4-二叔丁基咔唑亚丙氧基苯基苯并咪唑溶解于四氢呋喃中,加入1mmol的(4-3-氯丙氧基)苯基二苯基膦氧、0.3mmol的tbab,2mmol的nah,70℃条件下搅拌反应20小时,然后用水和ch2cl2萃取,合并有机相,用无水硫酸钠干燥,以石油醚和乙酸乙酯的混合溶剂为淋洗剂柱层析纯化得到2,4-二叔丁基咔唑亚丙氧基苯基-三苯基膦氧亚丙基苯并咪唑;
三、将步骤二合成的配体2,4-二叔丁基咔唑亚丙氧基苯基-三苯基膦氧亚丙基苯并咪唑、0.8mmol的三氯化铱、12ml的乙二醇独乙醚、12ml的水以及8ml的四氢呋喃加热至110℃搅拌反应20小时。然后冷却至室温,抽滤得到氯桥联中间体。将此中间体与1当量的2,4-二叔丁基咔唑亚丙氧基苯基-三苯基膦氧亚丙基苯并咪唑、3当量的k2co3以及10ml甘油加热至240℃搅拌反应24小时。反应结束后用水和ch2cl2萃取,合并有机相,用无水硫酸钠干燥,以石油醚和乙酸乙酯的混合溶剂为淋洗剂柱层析纯化得到铱配合物ir(24dcznpopbi)3。
本实验步骤一制备的2,4-二叔丁基咔唑亚丙氧基苯基苯并咪唑,其结构式为
按照实验二中步骤一制备的2,4-二叔丁基咔唑亚丙氧基苯基苯并咪唑,其飞行时间质谱的数据为:m/z(%):864(100)[m+];元素分析的数据为:分子式c59h68n4o2,理论值:c81.90,h7.92,n6.48,实测值:c81.92,h7.89,n6.49。
步骤二制备的2,4-二叔丁基咔唑亚丙氧基苯基-三苯基膦氧亚丙基苯并咪唑,其飞行时间质谱的数据为:m/z(%):1198(100)[m+];元素分析的数据为:分子式c80h87n4o4p,理论值:c80.10,h7.31,n4.67;实测值:c80.11,h7.36,n4.64。
步骤三制备的铱配合物ir(24dcznpopbi)3,其飞行时间质谱的数据为:m/z(%):3790(100)[m+];元素分析的数据为:分子式c240h261irn12o12p3,理论值:c76.04,h6.94,n4.43;实测值:c76.08,h6.90,n4.41。
本实验得到咔唑和膦氧双修饰均配位苯基苯并咪唑铱配合物ir(24dcznpopbi)3的紫外荧光光谱谱图如图3所示。
本实验得到咔唑和膦氧双修饰均配位苯基苯并咪唑铱配合物ir(24dcznpopbi)3的热重分析谱图如图4所示,由图可知铱配合物ir(24dcznpopbi)3的裂解温度达350℃。
实验三:本实验咔唑和膦氧双修饰均配位苯基苯并咪唑铱配合物ir(34dcznpopbi)3的合成方法按下列步骤实现:
一、将1mmol的3,4-二羟基苯基苯并咪唑、0.3mmol的tbab、3mmol的koh、3mmol的9-(3-氯丙基)叔丁基咔唑溶解于20ml四氢呋喃中70℃条件下搅拌反应20小时,然后用水和ch2cl2萃取,合并有机相,用无水硫酸钠干燥,以石油醚和乙酸乙酯的混合溶剂为淋洗剂柱层析纯化得到3,4-二叔丁基咔唑亚丙氧基苯基苯并咪唑;
二、将步骤一合成的3,4-二叔丁基咔唑亚丙氧基苯基苯并咪唑溶解于四氢呋喃中,加入2mmol的(4-3-氯丙氧基)苯基二苯基膦氧、0.3mmol的tbab,2mmol的nah,70℃条件下搅拌反应18小时,然后用水和ch2cl2萃取,合并有机相,用无水硫酸钠干燥,以石油醚和乙酸乙酯的混合溶剂为淋洗剂柱层析纯化得到3,4-二叔丁基咔唑亚丙氧基苯基-三苯基膦氧亚丙基苯并咪唑;
三、将步骤二合成的配体3,4-二叔丁基咔唑亚丙氧基苯基-三苯基膦氧亚丙基苯并咪唑、0.7mmol的三氯化铱、14ml的乙二醇独乙醚、14ml的水以及8ml的四氢呋喃加热至110℃搅拌反应22小时。然后冷却至室温,抽滤得到氯桥联中间体。将此中间体与2当量的3,4-二叔丁基咔唑亚丙氧基苯基-三苯基膦氧亚丙基苯并咪唑、3当量的k2co3以及10ml甘油加热至240℃搅拌反应30小时。反应结束后用水和ch2cl2萃取,合并有机相,用无水硫酸钠干燥,以石油醚和乙酸乙酯的混合溶剂为淋洗剂柱层析纯化得到铱配合物ir(34dcznpopbi)3。
本实验步骤一制备的3,4-二叔丁基咔唑亚丙氧基苯基苯并咪唑,其结构式为
按照实验三中步骤一制备的3,4-二叔丁基咔唑亚丙氧基苯基苯并咪唑,其飞行时间质谱的数据为:m/z(%):864(100)[m+];元素分析的数据为:分子式c59h68n4o2,理论值:c81.90,h7.92,n6.48,实测值:c81.92,h7.96,n6.51。
步骤二制备的3,4-二叔丁基咔唑亚丙氧基苯基-三苯基膦氧亚丙基苯并咪唑,其飞行时间质谱的数据为:m/z(%):1198(100)[m+];元素分析的数据为:分子式c80h87n4o4p,理论值:c80.10,h7.31,n4.67;实测值:c80.11,h7.35,n4.70。
步骤三制备的铱配合物ir(34dcznpopbi)3,其飞行时间质谱的数据为:m/z(%):3790(100)[m+];元素分析的数据为:分子式c240h261irn12o12p3,理论值:c76.04,h6.94,n4.43;实测值:c76.00,h6.95,n4.48。
本实验得到咔唑和膦氧双修饰均配位苯基苯并咪唑铱配合物ir(34dcznpopbi)3的紫外荧光光谱谱图如图5所示。
本实验得到咔唑和膦氧双修饰均配位苯基苯并咪唑铱配合物ir(34dcznpopbi)3的热重分析谱图如图6所示,由图可知铱配合物ir(34dcznpopbi)3的裂解温度达336℃。
将制得的铱配合物ir(34dcznpopbi)3和tpxzpo的掺杂体作为发光层制备掺杂型有机电致磷光器件。具体制备方法按以下步骤实现:
发光层为铱配合物ir(34dcznpopbi)3和tpxzpo的掺杂体,旋涂成膜,厚度为40nm.在阳极(氧化铟锡ito)和发光层之间旋涂上一层厚度为30nm的空穴注入层(聚3,4-乙二氧基噻吩:聚乙烯磺酸盐,pedot:pss)。电子传输/空穴阻挡层所用材料为1,3,5-三[(3-吡啶基)-3-苯基]苯(tmpypb),薄膜厚度为40nm。电子注入层在阴极和电子传输/空穴阻挡层之间,所用材料为lif,厚度为1nm。电极材料为铝,厚度为100nm,电子传输/空穴阻挡层、电子注入层和电极材料均采用真空热蒸发技术涂膜。器件的结构为ito/pedot:pss(30nm)/tpxzpo:ir(34dcznpopbi)3(40nm)/tmpypb(40nm)/lif(1nm)/al(100nm)
本实验以铱配合物ir(34dcznpopbi)3制备的电致绿光磷光器件的电压-电流密度关系曲线如图7所示,由此图可知铱配合物ir(34dcznpopbi)3具有半导体特性,其阀值电压为3.5v。
本实验以铱配合物ir(34dcznpopbi)3制备的电致绿光磷光器件的电压-亮度关系曲线如图8所示,由此图可知该器件的最大亮度可达12820cd·m-2。
本实验以铱配合物ir(34dcznpopbi)3制备的电致绿光磷光器件的电流密度-电流效率关系曲线如图9所示,由此图可知该器件在电流密度为0.18ma·cm-2时,电流效率达到最大值35.6cd·a-1。
本实验以铱配合物ir(34dcznpopbi)3制备的电致绿光磷光器件的电流密度-功率效率关系曲线如图10所示,由此图可知该器件在电流密度为0.04ma·cm-2时,功率效率达到最大值22.7lm·w-1。
本实验以铱配合物ir(34dcznpopbi)3制备的电致深蓝光磷光器件的电流密度-外量子效率关系曲线如图11所示,由此图可知该器件在电流密度为0.18ma·cm-2时,获得最大外量子效率10.4%。
本实验以铱配合物ir(34dcznpopbi)3制备的电致绿光磷光器件的电致发光光谱图如图12所示,由此图可知该器件的电致发光峰在525nm处,伴随560nm的肩峰。
本实验咔唑和膦氧双修饰均配位苯基苯并咪唑铱配合物ir(34dcznpopbi)3制备的非掺杂型电致绿光磷光器件按以下方法制备:
发光层为铱配合物ir(34dcznpopbi)3,旋涂成膜,厚度为40nm。其余各层材料和成膜方法与实验四相同。器件的结构为ito/pedot:pss(30nm)/ir(34dcznpopbi)3(40nm)/tmpypb(40nm)/lif(1nm)/al(100nm)
本实验以铱配合物ir(34dcznpopbi)3制备的非掺杂型电致绿光磷光器件的电压-电流密度关系曲线如图13所示,由此图可知铱配合物ir(34dcznpopbi)3具有半导体特性,其阀值电压为3.0v。
本实验以铱配合物ir(34dcznpopbi)3制备的非掺杂型电致绿光磷光器件的电压-亮度关系曲线如图14所示,由此图可知该器件的最大亮度可达16940cd·m-2。
本实验以铱配合物ir(34dcznpopbi)3制备的非掺杂型电致绿光磷光器件的电流密度-电流效率关系曲线如图15所示,由此图可知该器件在电流密度为0.005ma·cm-2时,电流效率达到最大值44.1cd·a-1。
本实验以铱配合物ir(34dcznpopbi)3制备的非掺杂型电致绿光磷光器件的电流密度-功率效率关系曲线如图16所示,由此图可知该器件在电流密度为0.005ma·cm-2时,功率效率达到最大值46.2lm·w-1。
本实验以铱配合物ir(34dcznpopbi)3制备的非掺杂型电致绿光磷光器件的电流密度-外量子效率关系曲线如图17所示,由此图可知该器件在电流密度为0.005ma·cm-2时,获得最大外量子效率12.5%。
本实验以铱配合物ir(34dcznpopbi)3制备的非掺杂型电致绿光磷光器件的电致发光光谱图如图18所示,由此图可知该器件的电致发光峰在527nm处,伴随560nm的肩峰。
- 还没有人留言评论。精彩留言会获得点赞!