法尼基焦磷酸合成酶条件性敲除模型构建方法和用途与流程
本发明属于生物技术领域,具体涉及一种法尼基焦磷酸合成酶基因条件性敲除小鼠模型构建方法和用途。
背景技术:
随着人类基因组计划的完成,生命科学研究进入了后基因组时代,而目前主要的研究工作就是对基因的功能进行深入的研究。目前,研究基因功能最常用的技术就是对靶向基因进行人工干预,即基因打靶技术,包括基因敲除、基因敲入、基因点突变等。通常意义上的基因敲除主要是应用dna同源重组原理,用设计的同源片段替代靶基因片段,从而达到基因敲除的目的。随着基因敲除技术的发展,除了同源重组外,新的原理和技术也逐渐被应用,它们同样可以达到基因敲除的目的。
法尼基焦磷酸合成酶(fdps)是类异戊二烯生物合成的关键酶,它催化异戊烯基焦磷酸(ipp)和二甲丙烯基焦磷酸(dmapp)形成中间产物牻牛儿基焦磷酸(gpp)和终产物法尼基焦磷酸(fpp)。fpp是胆固醇和固醇形成的重要中间物,另外,fpp也是牻牛儿牻牛儿基焦磷酸(ggpp)合成的底物。fpp和ggpp在蛋白质法尼基化和牻牛儿牻牛儿基化过程中起重要作用,包括小g蛋白,如rhoa,ras等。法尼基化和牻牛儿牻牛儿基化是小g蛋白活化的必要环节。
fdps完全性基因敲除小鼠有可能导致胚胎致死,因为fdps基因在胚胎期高表达。条件性基因敲除小鼠是通过基因打靶的策略,在靶基因特定区域两端插入方向相同的loxp位点,当该小鼠在导入cre重组酶的情况下(如和某组织特异性表达cre的转基因小鼠交配或者利用病毒介导等方式),cre酶通过识别loxp位点,切除两个同方向loxp位点间的序列,达到在特定时间或特定细胞中条件性敲除基因的目的,降低小鼠自发性死亡的可能性。
技术实现要素:
本发明的目的是提供一种法尼基焦磷酸合成酶(fdps)基因条件性敲除小鼠模型构建方法,具体通过以下方法实现:
1.构建fdps条件性基因敲除打靶载体;
2.线性化重组载体转染小鼠胚胎干细胞;
3.筛选正确的同源重组克隆;
4.将正确的同源重组克隆注射供体小鼠囊胚;
5.将上一步中的囊胚移植到假孕小鼠子宫,生产得到嵌合体小鼠;
6.嵌合体小鼠与flp小鼠回交得到fdps-flox杂合子小鼠;
7.fdps-flox杂合子小鼠繁育得到fdps-flox纯合子小鼠。
根据本发明优选的,步骤1构建fdps条件性基因敲除重组载体步骤如下:
(1)提取含有fdps基因的细菌人工染色体(bac);
(2)以bac为模板dna,根据表1设计引物,分别pcr扩增a,b,c,d4个片段;以c1和d2为引物,c和d为模板dna,pcr扩增得到e片段,大小为725bp;
表1
(3)用sali和clai酶切a片段,clai和ecori酶切b片段,sali和ecori酶切pl-451质粒,将上述片段连接后质粒转化感受态细菌,涂板,挑取单克隆后测序,鉴定出连接正确的命名为pl451-2。用sali和sacii酶切pl451-2,sacii和spei酶切e片段,sali和spei酶切pbr322-2s质粒,将上述三片段连接后质粒转化感受态细菌,涂板,挑取单克隆后测序,得到pfdps-1质粒;
(4)用xhoi线性化pfdps-1。以bac为模板,通过taqdna聚合酶同源重组的方法获得c与d之间的dna片段。获得的质粒命名为pfdps-cko;
(5)经过实验测试,使用上一步中的质粒载体无法获得纯合子小鼠,我们对打靶质粒进行了修正。以bac质粒为模板dna,pcr扩增新敲除区域g;以上一步中pfdps-cko质粒为模板dna,pcr扩增打靶载体骨架h。g和h引物见表2;
表2
(6)将g片段和h载体进行in-fusion反应,得到新质粒pfdps-cko2。该质粒即为本发明最终优选出的fdps条件性基因敲除打靶载体。
本发明的第二个目的是提供所述的构建方法在心血管重塑研究中的应用。将本发明获得的fdps-flox纯合子小鼠与心肌特异性表达cre重组酶的myh6-mercremer小鼠交配获得在心肌特异性表达cre重组酶的fdpsfl/flcre+/-小鼠,给予他莫昔芬后即可获得时间调控的心肌组织特异性敲除fdps的小鼠。
本发明首次获得具有fdps基因条件性敲除的小鼠模型。由于fdps基因有两个转录本,通过本发明优选的打靶载体可以同时影响这两个转录本,使这两个转录本均无法正确翻译成fdps蛋白,达到完全敲除的效果。本发明的有益之处在于可以选择性的在特定时间和特定组织内敲除fdps蛋白,避免了全身敲除导致的小鼠胚胎致死的可能性。
附图说明
图1是打靶载体设计图。外显子编号根据fdps-202转录本。
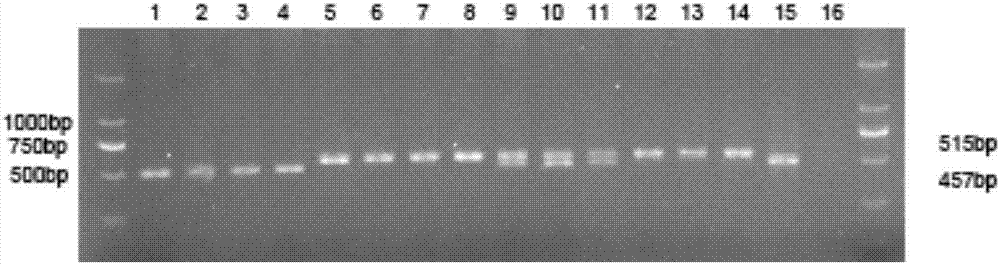
图2是flox小鼠基因型鉴定。
图3是心肌特异性敲除fdps蛋白水平验证。
具体实施方式
下面结合实施例与附图对本发明的技术方案做进一步说明,但本发明所保护范围不限于此。
实施例1fdps条件性基因敲除小鼠的构建
1.构建小鼠fdps基因打靶载体。
本载体设计是将小鼠fdps基因翻译起始位点序列敲除,从而使fdps蛋白无法翻译。首先从ensembl数据库中(http://www.ensembl.org/index.html)获得fdps基因组序列,根据基因组序列设计打靶载体。小鼠fdps基因fdps-201转录本共有10个外显子,根据生物信息学分析,flox区域为含有翻译起始位点atg的第2外显子,loxp、neo、frt等元件放在内含子中,flox区域敲除后可造成翻译无法起始,无蛋白生成。构建fdps基因第2外显子两侧插入loxp位点的条件性敲除载体。具体方法如下:
(1)提取含有fdps基因的细菌人工染色体(bac,购自sourcebioscience公司)。
(2)以bac为模板dna,根据表1设计引物,分别pcr扩增a,b,c,d4个片段。以c1和d2为引物,c和d为模板dna,pcr扩增得到e片段,大小为725bp。
表1
(3)用sali和clai酶切a片段,clai和ecori酶切b片段,sali和ecori酶切pl-451质粒,将上述片段连接后质粒转化感受态细菌,涂板,挑取单克隆后测序,鉴定出连接正确的命名为pl451-2。用sali和sacii酶切pl451-2,sacii和spei酶切e片段,sali和spei酶切pbr322-2s质粒,将上述三片段连接后质粒转化感受态细菌,涂板,挑取单克隆后测序,得到pfdps-1质粒。
(4)用xhoi线性化pfdps-1。以bac为模板,通过taqdna聚合酶同源重组的方法获得c与d之间的dna片段。获得的质粒命名为pfdps-cko。
之后的实验中,我们发现除了fdps-201外,还有fdps-202转录本。fdps-202转录本共有11个外显子,根据生物信息学分析,该转录本比fdps-201多出一个含有翻译起始位点外显子。我们对打靶质粒进行了修正,将这两个转录本的翻译起始位点一起敲除。具体方式如下:
(1)以bac质粒为模板dna,pcr扩增新敲除区域g;以上一步中pfdps-cko质粒为模板dna,pcr扩增打靶载体骨架h。引物序列如表2。
表2
(2)将g片段和h载体进行in-fusion反应,得到新质粒pfdps-cko2。
in-fusion反应体系如下:
(3)将反应体系置于50℃孵育15min,然后置于冰上。将pfdps-cko2质粒转化感受态细菌,涂板,挑取单克隆后测序,确定序列正确。pfdps-cko2质粒即为本发明最终优选出的fdps条件性基因敲除打靶载体。
2.载体质粒线性化
pfdps-cko2载体质粒用noti单酶切线性化后再进行纯化,用适量无水乙醇洗涤,加入适量te溶解质粒,用nanodrop测量dna浓度,调整至1μg/μl备用。
3.es细胞培养
培养皿以0.1%gelatin包被后接种丝裂霉素c处理的滋养层细胞(每个10cm盘接种2×106个滋养层细胞),过夜培养后即可接种es细胞。es细胞完全培养基为dmem中含1×10-6mol/lβ-巯基乙醇,2mm谷胺酰胺,0.1mm非必须氨基酸,100u/ml青霉素,50mg/l链霉素,15%es胎牛血清,1000u/mllif。
4.es细胞电穿孔转移线性化质粒
取处于对数生长期的es细胞,加0.125%胰蛋白酶-edta消化并计数,加适量的pbs使细胞密度达到约1.5×107/m1。取0.8ml上述es细胞悬液,加入约35μg步骤2中线性化的质粒dna,混匀后转移至无菌电穿孔杯中,轻轻吹打混匀。以240v、500μf的电参数进行电穿孔,实际电压和放电时间分别为256v、10.6ms。立即将电转杯置于冰上,将细胞重新悬浮后平均分配到三个已铺好滋养层细胞的10cm盘培养皿中培养。
5.正负药物筛选
es细胞在电穿孔24h和48h后分别换含有选择药物g418(终浓度为300mg/l)和ganciclovoir(终浓度为2umol/l)的培养液进行选择性培养。每天更换培养液,经7-8天选择性培养,抗性es细胞长成肉眼可见的克隆时即可进行挑取。
6.双抗性细胞克隆的挑取及培养
挑取抗性克隆,放入含有30μl0.1%胰蛋白酶-edta的96孔板(凹底)中消化约3min,轻轻吹打,使细胞分散。转移至96孔培养板中培养,待细胞长满到60%-80%后取大部分细胞冻存,剩余细胞继续培养至长满100%后用于提取基因组dna。
7.es细胞基因组dna的提取
在长满es细胞的96孔板中吸去培养液,每孔加入80μl裂解液(含1g/l蛋白酶k),56℃过夜消化后加入无水乙醇常规提取dna,溶于100μlte中。
8.同源重组克隆的pcr鉴定
如图1所示,在fdps打靶同源重组臂外侧分别设计p1和p4引物,在neo序列上设计p2和p3引物。序列如下:
p1:5′-tctgcgagccatgttcaatctac-3′
p2:5′-ctgagcccagaaagcgaagga-3′
p3:5′-gtgccactcccactgtcctttcc-3′
p4:5′-ccccggtagcaggagagtgtaaaa-3′
应用lataq(takara)进行pcr扩增,体系如下:
用p1与p2引物配对,鉴定5′臂同源重组,pcr扩增条件为:
同源重组克隆可扩增出2.8kb片段;用p3和p4引物配对鉴定3′臂同源重组,反应条件为:
同源重组克隆可扩增出5.0kb片段。以上pcr产物经1%琼脂糖凝胶电泳120v,20min。凝胶成像系统分析扩增产物条带。阴性组没有特定长度的条带。将同时拥有5′臂和3′臂的阳性克隆胶回收,测序。本例中,我们检测到有6个可能正确的同源重组克隆,分别为第b5,b6,c1,f4,f11,g6号克隆。通过胶回收试剂盒回收各个阳性克隆的pcr产物,经过酶切、测序确认,表明b5,c1,f4,g6号胚胎干细胞克隆为正确同源重组克隆。
9.es细胞的囊胚注射及胚胎移植
将上一步中鉴定正确的es细胞,注入小鼠囊胚中。注射时采用的是无lif的dmem完全培养基,每枚囊胚注射15个左右的es细胞。注射后囊胚培养于无lif的dmem完全培养基中,37℃,5%co2条件下培养1h左右。将注射后的胚胎移植到2.5天假孕母鼠子宫中,每侧移植8-10枚。待其自然分娩,产下的后代即为嵌合体小鼠。本例中,我们共注射146枚胚胎,植入12只假孕母鼠,共出生7只雄鼠,其中5只为>50%嵌合雄鼠。
10.纯合子小鼠育种及pcr鉴定小鼠基因型
挑选毛色嵌合率大于50%的雄性鼠与flp小鼠交配即获得es细胞来源的仔鼠。在fdps载体neo的上游及下游的野生型基因组上分别设计p5和p6引物。引物序列如下:
p5:5′-agagcatttagctcctctgtc-3′
p6:5′-caaacatccccgaggagggctgaag-3′
小鼠编号后提取亲本和仔鼠的鼠尾组织dna,步骤如下:
(1)无菌剪取5mm鼠尾组织,剪碎,加入300μl组织裂解液(nacl100mmol/ledta0.5mmol/ltris-hcl100mmol/l)。加入150μg蛋白酶k,55℃水浴过夜。
(2)消化产物室温10000rpm离心10min。
(3)取上清,加入等体积tris饱和酚,颠倒混匀,10000rpm离心5min。
(4)取上清,加入等体积饱和酚/氯仿/异戊醇(25:24:1)抽提,待分层后10000rpm离心5min。
(5)取上层水相,加等体积氯仿抽提,颠倒混匀,10000rpm离心5min。
(6)取上层水相,加入1/10体积3m醋酸钠和2倍体积无水乙醇,颠倒混匀,放入-20℃沉淀dna1-2h。
(7)12000rpm离心15min。去上清,加入70%乙醇500μl洗涤沉淀,12000rpm离心5min。(8)室温干燥dna沉淀,溶于100μlte溶液或者无菌去离子水中。
将小鼠dna提取液1μl,预混2×pcrmix(康为世纪)10μl,p5引物和p6引物各0.4μl,ddh2o8.2μl混匀后,pcr扩增,条件为:
pcr产物经1%琼脂糖凝胶电泳120v电泳分离20min,凝胶成像系统分析扩增产物条带。野生型小鼠只能得到298bp条带,去neo杂合子小鼠同时含有298bp和454bp的条带。将454bp条带胶回收测序。本例中来自g6克隆的1只雄鼠共获得es细胞来源的灰鼠16只。经pcr鉴定其中5只为阳性杂合子小鼠(1雄4雌)。
将雄性和雌性去neo杂合子小鼠(flox+/-)交配,从子代中筛选flox纯合子(flox+/+)。鉴定flox序列使用p7和p8引物,条件为95℃2min变性后,95℃30s,66℃30s,72℃45s进行35个循环,72℃5min。pcr产物经1%琼脂糖凝胶电泳120v电泳分离20min,凝胶成像系统分析扩增产物条带。如附图2野生型小鼠只能得到457bp条带(flox-/-),杂合子小鼠同时含有457bp和515bp的条带(flox+/-),纯合子小鼠只能得到515bp条带(flox+/+)。该纯合子小鼠即为本例构建的fdps条件性敲除小鼠模型。
p7:5′-cgggacagatgacaagcg-3′
p8:5′-aggagcaagcaccgaagg-3′。
实施例2组织特异性敲除小鼠繁育
将实施例1中和纯合子小鼠(flox+/+)与组织特异性表达cre重组酶的cre工具鼠交配,筛选表达cre重组酶的杂合子子代后再与纯合子小鼠交配,即可鉴定出组织特异性敲除fdps基因的小鼠。此外,将cre基因重组于病毒中,通过特异性病毒引入纯合子小鼠基因组达到时间和(或)组织(区域)特异性基因敲除。
本例中,我们将纯合子小鼠(flox+/+)与心肌特异性表达cre重组酶的myh6-mercremer小鼠(flox-/-cre+/-)交配,从子代中筛选表达cre重组酶的flox杂合子(flox+/-cre+/-)。将flox+/-cre+/-小鼠与纯合子小鼠(flox+/+)交配,子代中可以筛选出flox+/+cre+/-小鼠,即为纯合敲除小鼠。筛选出flox+/+cre+/-小鼠与flox+/+纯合子小鼠交配,其子代即可稳定获得flox+/+cre+/-小鼠与flox+/+纯合子小鼠。
鉴定cre序列使用cref和crer引物,条件为95℃2min变性后,95℃30s,60℃30s,72℃30s进行35个循环,72℃5min。pcr产物经1%琼脂糖凝胶电泳120v电泳分离20min,凝胶成像系统分析扩增产物条带。阳性小鼠可以得到309bp的条带,阴性小鼠没有条带。
cref:5′-atttgcctgcattaccggtcg-3′
crer:5′-cagcattgctgtcacttggtc-3′
通过腹腔注射他莫昔芬诱导cre阳性小鼠敲除,与同窝cre阴性小鼠作对照,判断敲除的效果。具体方法为:取10周龄小鼠,用玉米油/无水乙醇(9:1)配置他莫昔芬10mg/ml,60mg/kg/d连续5天腹腔注射。注射最后一天起8周后取小鼠心肌组织,提取蛋白,免疫蛋白印迹法分析fdps表达变化。如附图3,分析结果显示,1至4为敲除组,5、6为对照组,敲除小鼠fdps表达明显降低。
<110>浙江大学
<120>法呢醇焦磷酸合成酶条件性敲除模型构建方法和用途
<160>22
<210>1
<211>32
<212>dna
<213>人工序列
<223>fdps敲除打靶载体片段a上游引物序列
<400>1
catgtcgacactctgtgagttgaagtttagcc32
<210>2
<211>54
<212>dna
<213>人工序列
<223>fdps敲除打靶载体片段a下游引物序列
<400>2
catatcgatctcctcctggggccttctcaactgaggactggacacccatatgcc54
<210>3
<211>66
<212>dna
<213>人工序列
<223>fdps敲除打靶载体片段b上游引物序列
<400>3
catatcgatataacttcgtataatgtatgctatacgaagttatgggggcctggcacagtgcccgct66
<210>4
<211>31
<212>dna
<213>人工序列
<223>fdps敲除打靶载体片段b下游引物序列
<400>4
catgaattcgagacccatctactccagagac31
<210>5
<211>32
<212>dna
<213>人工序列
<223>fdps敲除打靶载体片段c上游引物序列
<400>5
catccgcggtgctgcacttcagccctcctcgg32
<210>6
<211>32
<212>dna
<213>人工序列
<223>fdps敲除打靶载体片段c下游引物序列
<400>6
gcagtgctcgagaaggctgaggtggggtttgt32
<210>7
<211>32
<212>dna
<213>人工序列
<223>fdps敲除打靶载体片段d上游引物序列
<400>7
agccttcatgagcactgcagcagacagctgtg32
<210>8
<211>30
<212>dna
<213>人工序列
<223>fdps敲除打靶载体片段d下游引物序列
<400>8
catactagtcagtaagtgactgtgttggtg30
<210>9
<211>74
<212>dna
<213>人工序列
<223>fdps敲除打靶载体片段g上游引物序列
<400>9
aggccccaggaggagataacttcgtataatgtatgctatacgaagttatacaaacttttcatttcctgtgtgtt74
<210>10
<211>37
<212>dna
<213>人工序列
<223>fdps敲除打靶载体片段g下游引物序列
<400>10
tgtgccaggcccccaggactggacacccatatgccag37
<210>11
<211>19
<212>dna
<213>人工序列
<223>fdps敲除打靶载体片段h上游引物序列
<400>11
tgggggcctggcacagtgc19
<210>12
<211>46
<212>dna
<213>人工序列
<223>fdps敲除打靶载体片段h下游引物序列
<400>12
ctcctcctggggccttctcaactgcggagtgactgagatccaatag46
<210>13
<211>23
<212>dna
<213>人工序列
<223>fdps同源重组臂5’端鉴定上游引物序列
<400>13
tctgcgagccatgttcaatctac23
<210>14
<211>21
<212>dna
<213>人工序列
<223>fdps同源重组臂5’端鉴定下游引物序列
<400>14
ctgagcccagaaagcgaagga21
<210>15
<211>23
<212>dna
<213>人工序列
<223>fdps同源重组臂3’端鉴定上游引物序列
<400>15
gtgccactcccactgtcctttcc23
<210>16
<211>24
<212>dna
<213>人工序列
<223>fdps同源重组臂3’端鉴定下游引物序列
<400>16
ccccggtagcaggagagtgtaaaa24
<210>17
<211>21
<212>dna
<213>人工序列
<223>neo基因上游引物序列
<400>17
agagcatttagctcctctgtc21
<210>18
<211>25
<212>dna
<213>人工序列
<223>neo基因下游引物序列
<400>18
caaacatccccgaggagggctgaag25
<210>19
<211>18
<212>dna
<213>人工序列
<223>loxp位点上游引物序列
<400>19
cgggacagatgacaagcg18
<210>20
<211>18
<212>dna
<213>人工序列
<223>loxp位点下游引物序列
<400>20
aggagcaagcaccgaagg18
<210>21
<211>21
<212>dna
<213>人工序列
<223>cre重组酶上游引物序列
<400>21
atttgcctgcattaccggtcg21
<210>22
<211>21
<212>dna
<213>人工序列
<223>cre重组酶上游引物序列
<400>22
cagcattgctgtcacttggtc21
- 还没有人留言评论。精彩留言会获得点赞!